
Abstract
The approval of proteasome inhibitors bortezomib and carfilzomib and the E3 ligase antagonist thalidomide and its analogs, lenalidomide and pomalidomide, validates the ubiquitin–proteasome pathway as a source of novel drugs for treating cancer and, potentially, a variety of devastating illnesses, including inflammation, cardiovascular disease, and neurodegenerative disease. All elements of this critical regulatory pathway—the proteasome itself, E3 ligases (which conjugate ubiquitin to target proteins), and deubiquitylating enzymes (which deconjugate ubiquitin, reversing ligase action)—are potential therapeutic targets, and all have been worked on extensively during the past decade. No deubiquitylase inhibitors or activators have yet progressed to clinical trial, however, despite compelling target validation and several years of high-throughput screening and preclinical development of hits by numerous pharmaceutical companies, biotechnology organizations, and academic groups. The appropriateness of deubiquitylases as therapeutic targets in many disease areas is reviewed, followed by evidence that selective inhibitors of these cysteine proteases can be discovered. Because the lack of progress in drug-discovery efforts with deubiquitylases suggests a need for improved discovery methodologies, currently available platforms and strategies are analyzed, and improved or completely novel, unrelated approaches are considered in terms of their likelihood of producing clinically viable effectors of deubiquitylases.
Introduction: The Ubiquitin Pathway, Deubiquitylating Enzymes (DUBs), and Disease
The ubiquitin pathway is a complex cellular system consisting of enzymes that conjugate the 76-amino-acid protein tag ubiquitin to and deconjugate it from target proteins, thereby regulating the protein’s cell content, compartmentation, and signaling.1–3 Many of the mechanistic aspects of the ubiquitin pathway have been elucidated; early on, it was determined that ubiquitin conjugation is performed by a series of enzymes ending with E3 ligases, and that deconjugation is performed by deubiquitylating enzymes (DUBs), also known as ubiquitin proteases or isopeptidases. The initial studies of ubiquitin focused on the cell content of its target proteins as determined by their relative rates of synthesis and degradation; most soluble cellular proteins are degraded in the proteasome, a multicomponent structure that receives (poly)ubiquitin tagged proteins and facilitates their degradation by component proteases ( Fig. 1 ).4,5 It is intuitive that the modulation of the content of critical cell proteins by inhibiting or activating ubiquitin pathway enzymes would have pharmacologic consequences, and the approval of the proteasome inhibitor bortezomib by the U.S. Food and Drug Administration in 2003 initiated the drug discovery era for the ubiquitin–proteasome pathway.6,7 Bortezomib and second-generation drugs, including the recently approved carfilzomib, inhibit the protein-degradation activities contained within the proteasome. Most of the soluble and many misfolded proteins are degraded in the proteasome as a means of regulating the proteins’ cellular levels and structural integrity (and, thus, their cellular activity), and inhibition of proteasomal protease activities triggers proapoptotic, antisurvival activities secondary to the buildup of proteins that would normally not accumulate in cells. 7 Proteasome inhibitors are active drugs, but their use is restricted somewhat by side effects, attributed to a lack of selectivity in the spectrum of proteasomal targets for degradation; in addition, the emergence of resistance to bortezomib is a significant limitation,8, 9 pointing to a need for novel targeted chemotherapeutic agents. Enzymes of the ubiquitin–proteasome pathway that act on limited numbers of substrates—including E3 ligases and DUBs ( Fig. 1 )—have been considered as potential targets for the discovery or design of novel drugs for the treatment of cancer and other diseases.3,10 The current and projected status of DUBs as targets for drug discovery will be considered here.
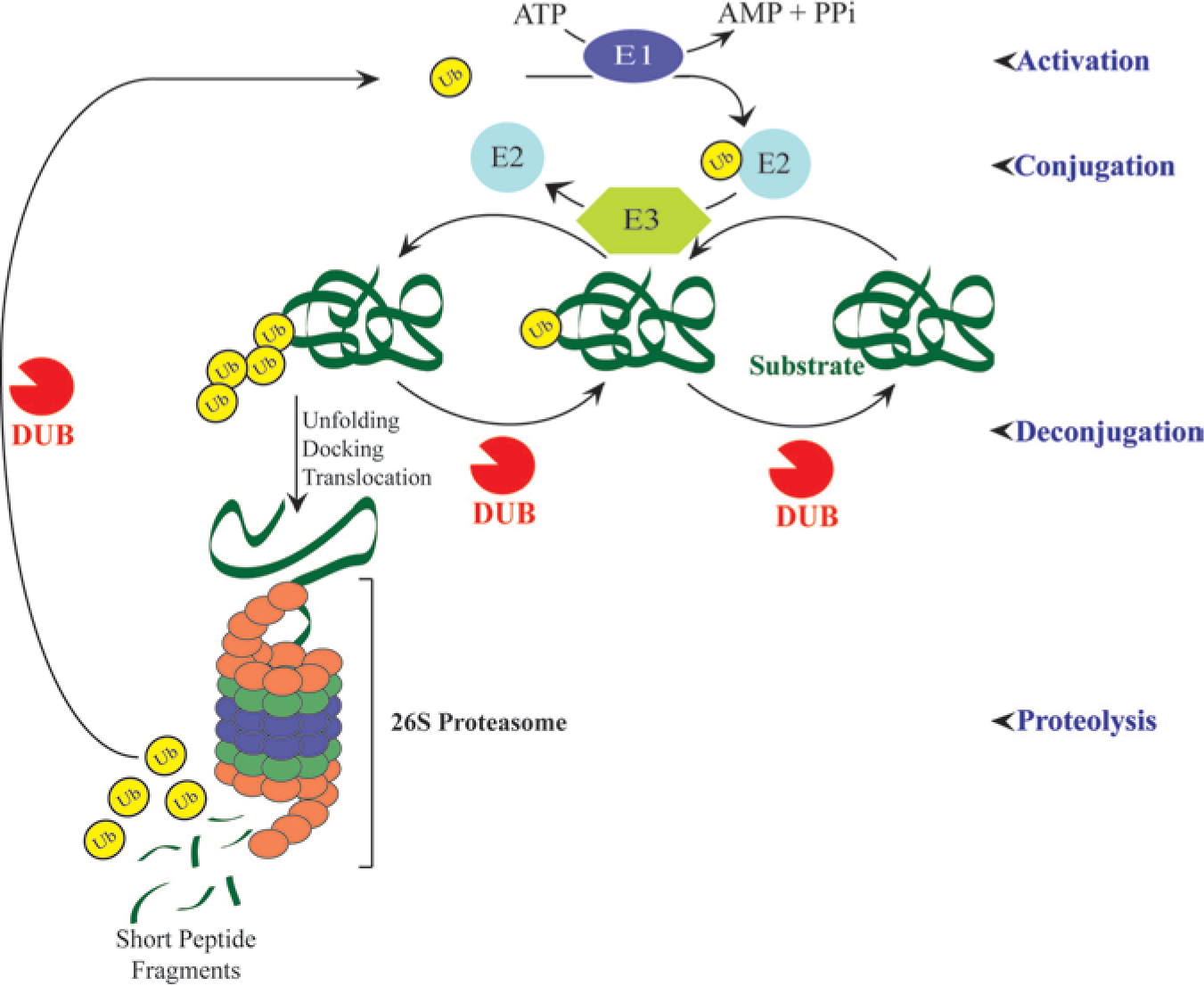
Ubiquitin–proteasome pathway. Protein targeting by ubiquitin (Ub) requires the sequential action of three enzymes: Ub is activated by activating enzyme (E1) to yield a Ub–E1–thioester. Activated Ub is transferred to “conjugase” E2, then transferred to a ligase (E3) and linked to an isopeptide bond of a lysine residue on the substrate protein. After linkage of Ub to the substrate, a poly-Ub chain is usually formed. Ubiquitylated proteins can be de-ubiquitylated by specific isopeptidases (deubiquitylating enzymes (DUBs)), or recognized and processed by the 26S proteasome. Proteasomes also contain DUB activity that allows Ub recycling, and vesicular structures associated with membranes contain DUBs that regulate endosomal sorting and recycling of receptors and other proteins to the membrane.
Approximately 80 DUBs, which hydrolyze isopeptide or α-peptide bonds linking ubiquitin to its target protein (in some cases, another ubiquitin), have been identified; each is presumed to have numerous potential substrates, but differences in levels of expression and cell physiology suggest that inhibition of a given DUB would not necessarily have global consequences. Many DUBs are validated targets for cancer and other diseases by cellular proof of concept and/or genetic evidence.11–14 Among their principal cellular functions are recycling ubiquitin monomers; preventing proteasomal degradation of proteins tagged with ubiquitin; trimming ubiquitin from tagged proteins in the proteasome; and mediating endocytosis and endosomal sorting of various signaling receptors, transporters, and channels.15–19 The sparing function (preventing certain proteins from being degraded in the proteasome) originally made DUBs attractive targets for cancer and other diseases. 18 If the DUB’s target protein is beneficial—for example, a tumor suppressor such as p53—DUB activity would be advantageous because it would increase its half-life. In this case, DUB activators would be sought. 13 In most cases, the opposite strategy—the use of DUB inhibitors to prevent the sparing of a deleterious target protein (e.g., an oncogenic protein)—has been pursued, primarily because it has been historically easier to find inhibitors of enzymes than activators. Two other physiological functions of DUBs—replenishment of ubiquitin pools, and regulating endosomal sorting or membrane recycling—are also of translational and potentially pharmacologic interest, and as more is learned about the nuances of ubiquitylation and deubiquitylation, as well as the multiple, complex ways in which they are regulated in cells, 20 additional therapeutic strategies likely will emerge.
DUBs Represent Attractive, Validated Therapeutic Targets for the Treatment of Numerous Diseases
A variety of DUBs have been implicated by biochemical and/or genetic evidence in most major diseases. The following are a few examples of DUBs that are currently being studied for drug development.13,15,21,22
USP7
The best-validated and most extensively exploited DUB target is arguably USP7 (originally named HAUSP owing to its herpesvirus association). USP7 (or HAUSP) is a cysteine protease identified originally as a binding partner for the herpes simplex viral (HSV) protein infected cell protein 0. 23 Several eukaryotic cellular proteins have been shown to bind USP7, and some of them are substrates of this DUB that play roles in cancer and other diseases.13,24 Primary USP7-related pharmaceutical targets are discussed throughout the remainder of this subsection.
MDM2 and HDM2
MDM2 and its human ortholog, HDM2, are E3 ligases that ubiquitylate the tumor suppressor p53, leading to its degradation; they also auto-ubiquitylate as part of a feedback mechanism. USP7 reverses auto-ubiquitylation of MDM2, prolonging the cell half-life of the oncoprotein MDM2 and thereby shortening the half-life of MDM2’s substrate, p53. 25 Inhibitors of USP7 would be expected to increase p53 activity and thereby exert an antitumor effect ( Fig. 2 ), and small-molecule inhibitors of USP7 have been in preclinical development as anticancer agents.26,27,28
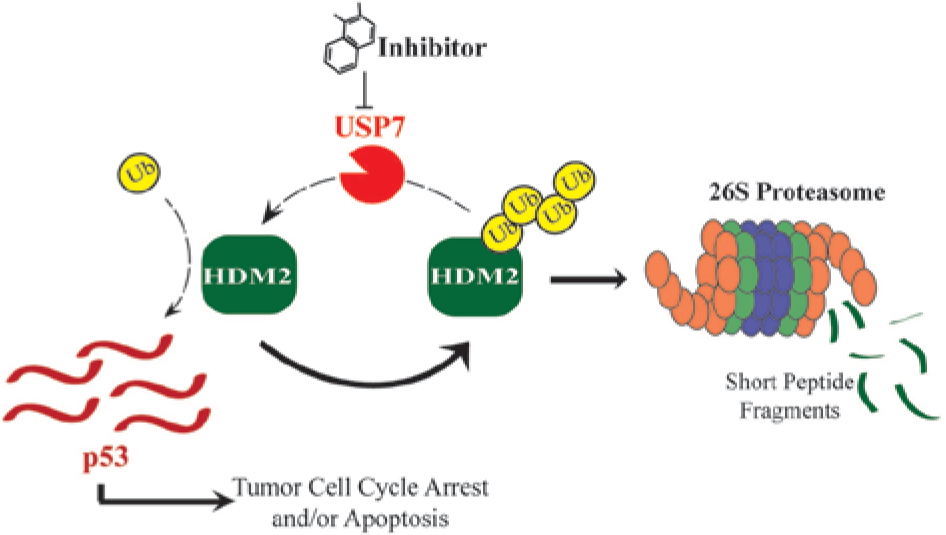
Inhibition of USP7 stabilizes the tumor suppressor p53. Inhibitors of USP7 render it incapable of stabilizing the E3 ligase HDM2, thereby leading to the degradation of HDM2 by the proteasome. One of the cellular consequences of the degradation of HDM2 is the stabilization of p53, which is then able to mediate antiproliferative effects on cancer cells. USP7 also mediates p53-independent effects such as the stabilization of claspin, a protein necessary for the DNA damage response.
Claspin
Claspin is a key adaptor protein that is required for checkpoint kinase 1 (Chk1) activation, which results in cell-cycle arrest under conditions of genotoxic stress, enabling the repair of DNA lesions. Inhibition of USP7 would be expected to attenuate or eliminate cell-cycle arrest in the presence of genotoxic chemotherapeutic agents, resulting in mitotic catastrophe and cell death. 29 USP7 inhibitors may be useful in combination with such agents in tumors irrespective of p53 status. 24
Foxp3
Foxp3 is a transcription factor that is required for the development of Treg cells, which suppress autoimmune responses. 30 USP7 deubiquitylates ubiquitylated Foxp3, sparing it from degradation and permitting suppression of T effector cells and facilitation of tumor growth. Thus, inhibition of USP7 is predicted to destabilize Foxp3, minimizing the immunosuppressive role of Tregs on T effector cells and potentially enabling immune-mediated tumor rejection. 31 This model suggests a novel mechanism, also p53 independent, whereby USP7 inhibitors could exert antitumor activity.
NFκB
USP7 is known to play a role in promoting pro-inflammatory cytokine expression through the nuclear factor kappa B (NFκB) pathway. Loss of USP7 activity results in increased ubiquitylation of NFκB, leading to reduced expression of genes induced by Toll-like receptor and tumor necrosis factor receptor activation. 32 USP7 inhibitors may therefore be useful as anti-inflammatory agents.
USP14
This DUB is the subject of intense interest in the search for new drugs to treat neurodegenerative and other neurological diseases. Associated with the 19S proteasome subunit, USP14 is required for synaptic function; Usp14 mutant mice (axJ) have a block in neuromuscular junction development and exhibit perinatal lethality. 33 In neurons, USP14 appears to perform an editing function, removing polyubiquitin chains from proteins and potentially rescuing them from degradation by the proteasome.34,35 Using a reconstituted purified USP14–proteasome model in screening, Finley and colleagues reported the first inhibitor of USP14, the compound IU1, 35 a selective inhibitor with an IC50 of ~5 µM. IU1 accelerates proteasome activity in vitro and enhances degradation of tau in a USP14-dependent manner in cellular models ( Fig. 3 ). 35 In addition, IU1 increases hydrolysis of other proteins that accumulate in neurodegenerative diseases, including TDP-43, ATXN3, and glial fibrillary acidic protein. 35 Thus, inhibition of USP14 indirectly enhances proteasome activity, increasing the degradation of tau and related proteins. This result is intriguing and has led to additional studies to exploit the observation for the development of new therapeutic agents.
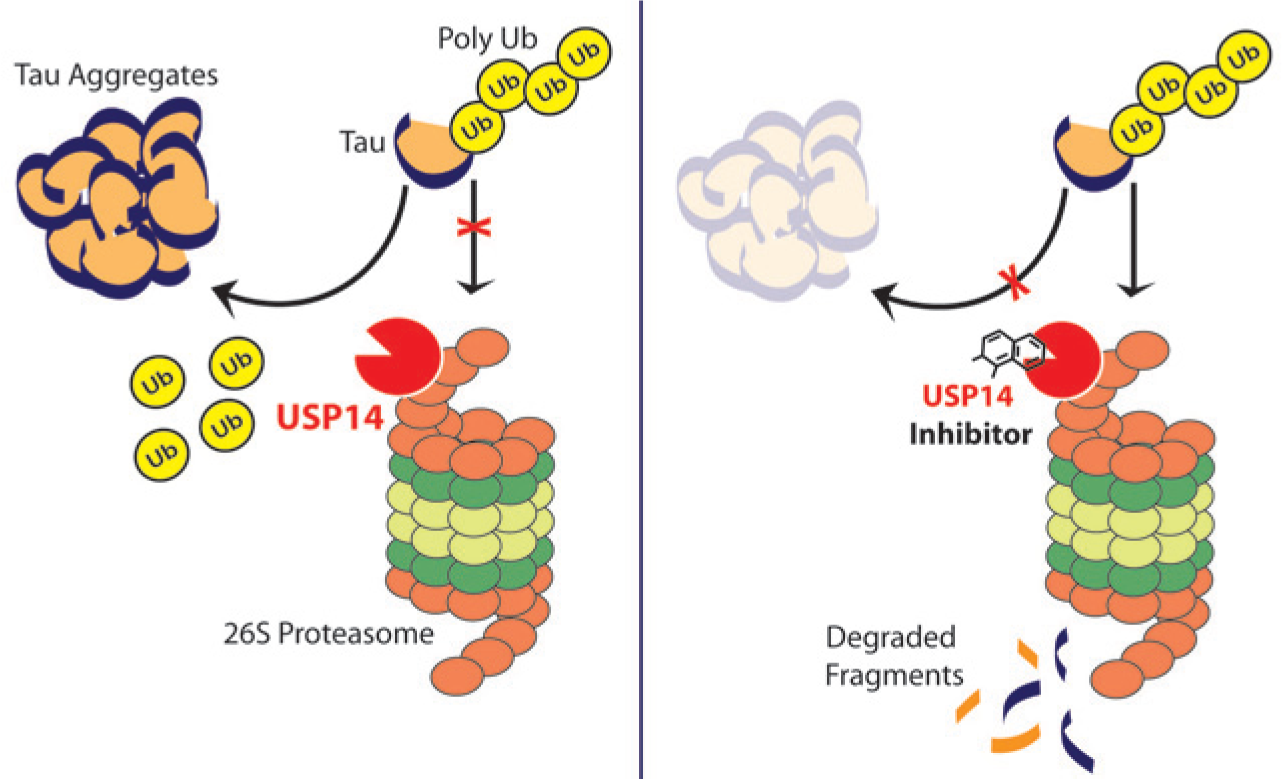
Effect of inhibiting USP14 in neurons. Left panel: Putative action of USP14 to deubiquitylate polyubiquitylated tau proteins in neurons, rescuing them from proteasomal degradation and favoring aggregation. Right panel: An inhibitor of USP14 (e.g., IU1) blocks this function, leading to diminished aggregate formation (see Lee et al. 19 ).
In addition, USP14 appears to regulate postsynaptic gamma-amino butyric acid-A receptor (GABAAR) density by rescuing the receptor from lysosomal degradation, favoring recycling of the GABAAR to the membrane. 36 In this setting, a USP14 inhibitor could increase GABAAR activity with a potential anxiolytic or sedative effect. Recent focusing on USP14 is a good example of the pharmaceutical industry’s keen interest in exploiting new targets to provide drugs to treat not only neurodegenerative diseases but also synapse-associated disorders with the aim of providing curative rather than symptomatic relief.
USP46
This DUB participates in receptor cycling in neuronal cells in a manner similar to that of USP14, negatively regulating the degradation of the glutamate receptor GLR-1 to control its density in the ventral nerve code of Caenorhabditis elegans.37,38 Changes in the abundance of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptors can alter the plasticity and strength of synaptic transmission, ultimately affecting central nervous system functions such as learning and memory. USP46 appears to deubiquitylate AMPA receptors (AMPARs), preventing degradation, in a fashion similar to the action of USP14 on GABAAR. Inhibition of USP46 should result in enhanced AMPAR ubiquitylation and degradation, limiting the intensity and duration of physiological responses. The role of USP46 in regulating human glutamate receptors is yet to be uncovered, however, so additional translational studies are necessary. One can speculate that USP46 inhibitors can increase the level of AMPAR at the postsynaptic surface, thus constituting a novel class of AMPAR activator with potential therapeutic benefits such as enhancement of cognitive function.
USP19
The ubiquitin–proteasome pathway plays a critical role in skeletal muscle protein breakdown under various stress conditions. Although the specific mechanisms involved in the tissue-specific regulation of muscle protein degradation are still largely unknown, USP19 is upregulated in skeletal muscle under conditions such as fasting, diabetes, dexamethasone treatment, and cancer, and is thus associated with muscle atrophy. 39 Both small interfering RNA and gene-knockout approaches that deplete USP19 protein have revealed significant sparing of specific myofibrillar proteins (myosin heavy chain, actin, troponin T, and tropomyosin) under atrophy conditions, in which they are disproportionately ubiquitylated and degraded. 40 In addition, USP19 appears to modulate transcription of myofibrillar proteins. These observations suggest that small-molecule inhibitors of USP19 could be used to prevent and/or slow down skeletal muscle wasting, a fatal complication of major diseases such as cancer, AIDS, and neuromuscular diseases. 41
Discovery of Selective Catalytic-Site Inhibitors of DUBs
DUBs are a family of proteases that specifically cleave ubiquitin-derived substrates of the general structure Ub-X, where X = a number of leaving groups ranging from small thiols and amines to Ub and other proteins.42,43 The majority of DUBs are cysteine proteases, grouped into four families—ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases (UCHs), ovarian tumor (OTU) domain proteases, and Josephin domain proteases. 14 DUBs are composed of catalytic domains as well as binding domains and protein–protein interaction domains. Because of these structural elements, various DUBs are able to bind to and recognize specific chain linkages and proteins that are important in determining a given DUB’s cellular locus and substrate, which govern its physiological activity.14,44 Cysteine proteases, sometimes referred to as thiol proteases, work by a common catalytic mechanism at the enzyme’s highly conserved active site. A thioester intermediate links the polypeptide substrate to the cysteine thiol, and hydrolysis of this thioester bond yields a deubiquitylated substrate.
The USPs comprise the largest class of DUBs and contain a conserved catalytic triad (Cys, His, and Asp) that forms the active site, illustrated in Figure 4 . The development of protease inhibitors as therapeutic agents is theoretically well justified, because proteases perform innumerable cellular functions that can be exploited pharmacologically. 3 Such development has been undertaken with caution, however, because in many instances the most potent inhibitory effects are achieved by molecules that interact with elements of this conserved active site (e.g., thiol-reactive compounds, which can be toxic). This prejudice has influenced attempts to discover inhibitors of DUBs, and the use of screening methodologies that depend on measurement of catalytic (hydrolysis) activity has resulted in very few molecules that are selective for one or a limited number of DUBs.26,28,45,46
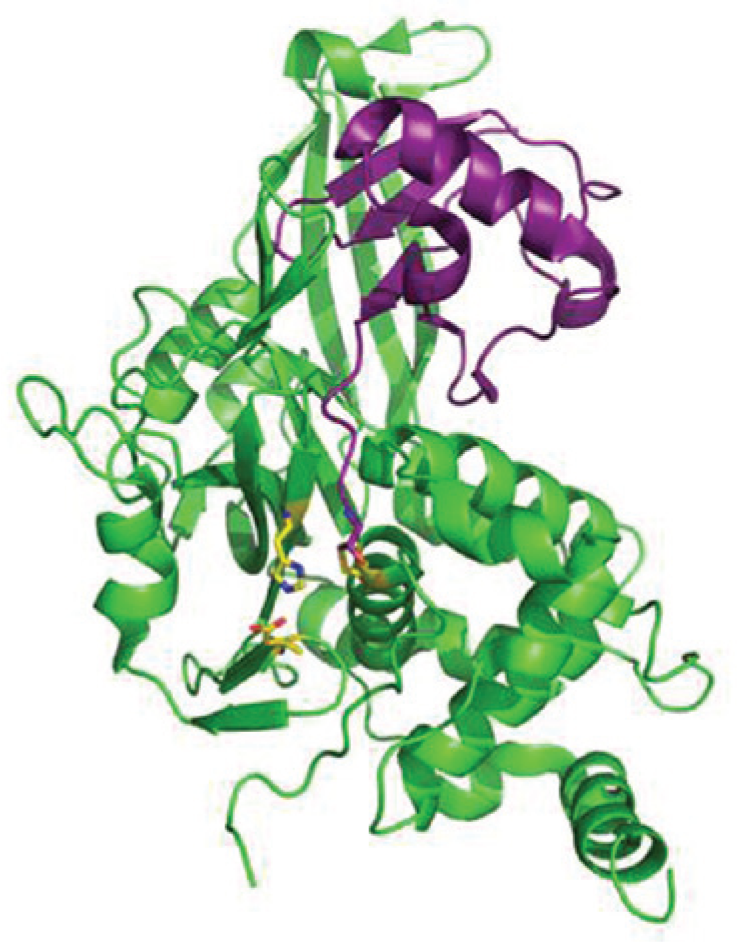
Structure of the catalytic core domain of USP7 with ubiquitin–aldehyde. The USP7 core is depicted in green with ubiquitin–aldehyde in purple. The conserved active site residues (catalytic triad: Cys, His, and Asp) are highlighted in yellow. The active site is in an unproductive conformation in the apo structure (pdb: 1NB8), 80 and on ubiquitin binding, the residues align for productive catalysis (pdb: 1NBF), 80 as shown.
Nevertheless, it is possible to find selective catalytic-site DUB inhibitors, some of which have demonstrated selectivity and in vivo activity ( Fig. 5 ). Pharmaceutical and academic researchers have reported on their efforts in this branch of target-based drug discovery. A group from AstraZeneca published results of their validation and deployment of a high-throughput screen for inhibitors of USP7, including an extensive biochemical characterization of the screen and resultant hits. 27 In high-throughput screening (HTS), Hybrigenics discovered HBX 41,108, a small-molecule compound that reversibly inhibits USP7 DUB activity with an IC50 in the submicromolar range and affects USP7-mediated p53 deubiquitiylation in a purified USP7 reaction system and in cells. The compound mimics USP7 RNA interference (RNAi)-mediated USP7 silencing in cancer cells—stabilizing p53, activating transcription of a p53 target gene without inducing genotoxic stress, inhibiting cancer cell growth, and inducing p53-dependent apoptosis. 26 More recently, a publication from Hybrigenics disclosed a second series of USP7 inhibitors, exemplified by HBX 19,818, that selectively inhibit USP7 by forming a covalent adduct with the active site Cys of USP7. 47 In the same publication, HBX 41,108 is described as a nonspecific DUB inhibitor. A different Hybrigenics compound, the DUB inhibitor 9-oxo-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile, gave rise to synthesized analogs that are potent and selective inhibitors of the DUB USP8. 46 The USP8 inhibitor reduced tumor growth in both gefitinib-sensitive and -resistant non-small-cell lung cancer xenografts. 48
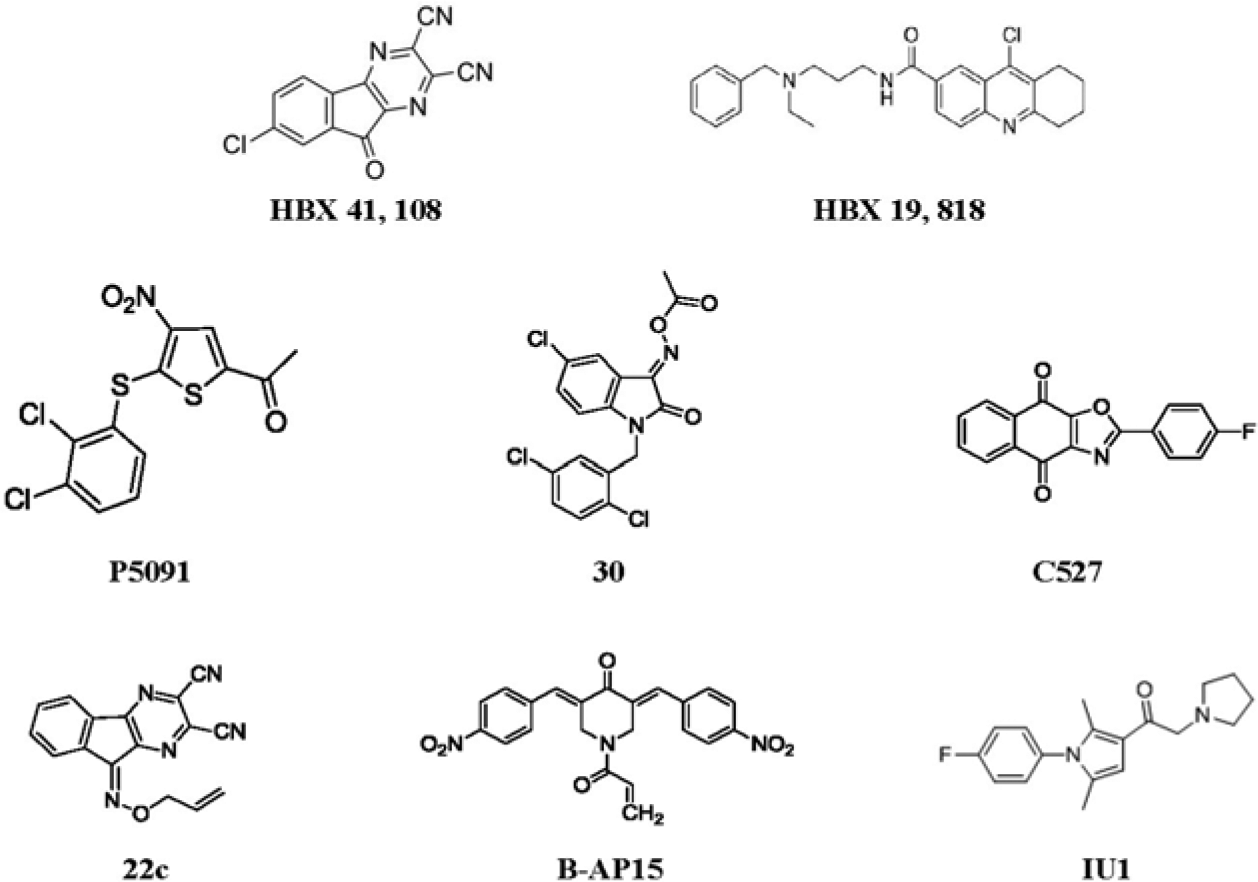
Selective experimental deubiquitylating enzyme (DUB) inhibitors. HBX 41,108, HBX 19,818, and P5091 inhibit USP7–HAUSP; IU1 is an inhibitor of USP14; C527 is an inhibitor of USP1; compound 30 inhibits UCH-L1; b-AP15 inhibits proteasome-associated DUBs UCH-L5 and USP14; and compound 22c inhibits USP8 (see the text under “Discovery of Selective Catalytic-Site Inhibitors of DUBs” for references).
At about the same time, Progenra reported a screening hit DUB inhibitor, P5091, which exhibits dual selectivity for USP7 and the closely related DUB USP47. P5091 and synthesized analogs exhibit moderate potency, demonstrate inhibition of USP7 in cells, and induce elevated p53 and apoptosis in cancer cell lines. P5091, in addition, significantly reduced tumor burden and significantly increased survival in four hematological malignancies.28,49,50 In these studies, inhibition of the target DUB in vivo was demonstrated. The activity of P5091 may be the result of inhibition of USP7 and USP47, both of which behave as oncoproteins. 28
Chen et al., working at the University of Delaware, identified selective small-molecule inhibitors of a DUB complex, human USP1–UAF1, through HTS. Bioactive molecules pimozide and GW7647 inhibit USP1–UAF1 with K(i) = 0.5 and 0.7 µM, respectively, and display selectivity against several DUBs, deSUMOylase, and other cysteine proteases. Moreover, the inhibitors synergize with cisplatin to inhibit growth of cisplatin-resistant non-small-cell lung cancer (NSCLC) cells. These results provide cellular proof of concept because USP1 is a key component of DNA damage repair and translesion synthesis.45,51
The small molecule b-AP15 is a proteasome inhibitor that blocks the deubiquitylating activity of two 19S regulatory-particle-associated deubiquitylases, ubiquitin C-terminal hydrolase 5 (UCHL5) and ubiquitin-specific peptidase 14 (USP14), resulting in an accumulation of polyubiquitin. 52 Treatment with b-AP15 has been shown to inhibit tumor progression in four different in vivo solid-tumor models and to inhibit organ infiltration in an acute myeloid leukemia model. In the solid-tumor animal models, it was shown also to reduce tumor burden and increase survival. 52
Finally, as noted above, Lee et al. reported the discovery, in high-throughput catalytic activity screening, of a small molecule, IU1, which inhibits the DUB USP14 selectively. 35 IU1 was subsequently licensed by Proteostasis Therapeutics as the basis for a drug-discovery program targeting Parkinson’s disease. IU1 and related compounds remain in preclinical development.
Although none of the aforementioned DUB inhibitors have entered clinical trials, their discovery serves to confirm the possibility of selective inhibition at the DUB catalytic site by a small molecule. Before considering other sites of inhibition on the DUB molecule that may be inhibited with increased potency and selectivity, the HTS assays historically and currently used to discover DUB inhibitors will be reviewed.
Methods Used to Discover DUB Inhibitors
These methods are summarized in Table 1 .
Summary of the Commercially Available Assays Used to Measure Deubiquitylating Enzyme (DUB) Activity in High Throughput.
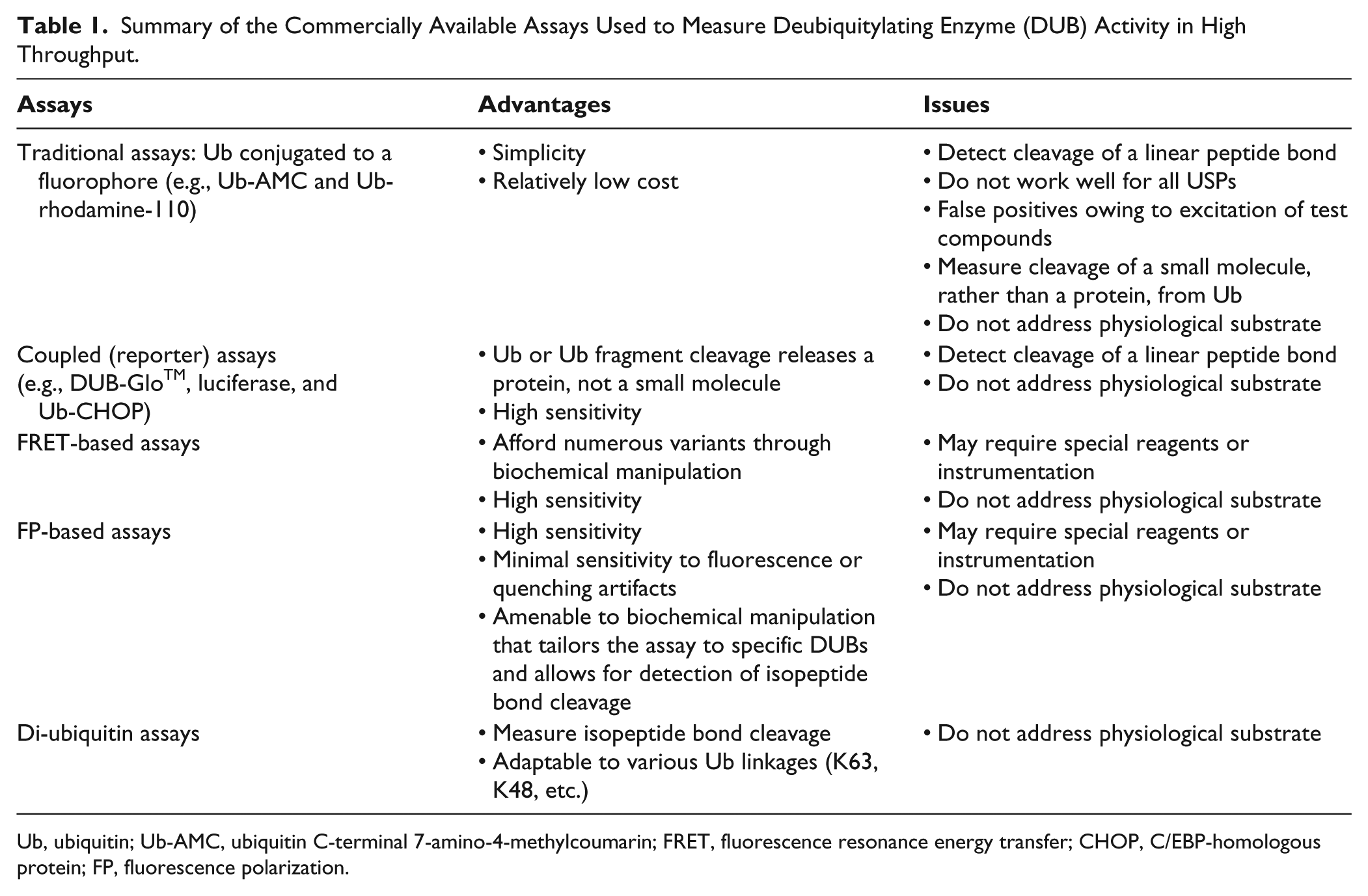
Ub, ubiquitin; Ub-AMC, ubiquitin C-terminal 7-amino-4-methylcoumarin; FRET, fluorescence resonance energy transfer; CHOP, C/EBP-homologous protein; FP, fluorescence polarization.
Traditional HTS Assays: Ubiquitin C-Terminal 7-Amino-4-Methylcoumarin (Ub-AMC) or Ub Conjugated to Alternative Fluorescent Moieties (e.g., Rhodamine-110)
The initial screening assays using these fluorogenic substrates measured the simple hydrolytic cleavage of a linear peptide bond between ubiquitin and a fluorescent organic molecule. Ub-AMC has been the mainstay. 53 A limiting factor with this and many other fluorescent substrates is the fact that this small adduct cannot be hydrolyzed efficiently by many Ub-specific protease (UBP) class enzymes, 54 limiting its utility in primary and selectivity screening. Moreover, AMC is highly hydrophobic and can interact with screening compounds; in addition, its excitation wavelength is in the ultraviolet range, which is known to excite a number of screening compounds and give rise to as many as 20% false positives. 55 Despite these drawbacks, AMC has been and continues to be widely used in HTS for DUB inhibitors.
Latter-Generation Assays Developed to Address Shortcomings of the Original Assays
The primary issues identified are fluorogenic substrate–related false positives; measurement of α−peptide rather than the isopeptide bond cleavage used by the majority of therapeutically interesting DUBs; and the measurement of cleavage of ubiquitin from a small molecule rather than a large polypeptide, its physiological substrate.
Reporter Assays
Several assays for DUB activity have been developed that entail ubiquitin (or a peptide fragment of the ubiquitin molecule) joined by a peptide bond to a reporter enzyme that is inactive when its amino terminus is bound to the carboxy terminus of ubiquitin (or a ubiquitin fragment). Deubiquitylase activity liberates the reporter enzyme with a free N-terminus, permitting it to act on its substrate to give a fluorescence-based signal. 56 This homogeneous assay is available in complete kit form or as components for the end user to incorporate into an assay tailored to a given DUB. If the excitation and emission wavelengths are appropriate, the coupled reporter assay can be run in multiplex format. 57
Promega markets the DUB-Glo™ Protease Assay, a homogeneous bioluminescence assay that measures the activity of numerous deconjugating enzymes, including DUBs. The assay provides a luminescent substrate, Z-RLRGG-aminoluciferin, a reagent optimized for protease and luciferase activity. The RLRGG sequence is the C-terminal pentapeptide of ubiquitin. A single DUB-Glo™ Reagent is added to test protease samples, resulting in substrate cleavage and generation of a luminescent signal produced by luciferase. In this coupled-enzyme format, luminescence is proportional to the amount of DUB activity present. Specificity resides in the protease cleavage sequence. Alternatively, attachment of D-amino-luciferin to the C-terminus of ubiquitin (and ubiquitin-like proteins) by a peptide bond results in a highly sensitive assay for certain DUBs for which no other HTS-amenable assay is efficacious. 58
Although the various coupled assays that are available for DUBs offer high sensitivity and/or the attachment of ubiquitin to a macromolecule of a size similar to that of the actual ubiquitin-conjugated DUB substrate, they do not measure isopeptidase activity or cleavage of the physiological substrate. The former shortcoming is significant in that not all DUBs hydrolyze linear peptide bonds. 59 In addition, the development of physiological substrate-based isopeptidase assays remains an important goal in ubiquitin-based drug discovery.
Fluorescence Polarization (FP) and Fluorescence Resonance Energy Transfer (FRET) Assays
A number of alternative ways to detect cleavage, such as FP 60 and FRET61,62 assays, have been developed into high-throughput screens for DUB activity. These methods typically require specialized custom reagents and equipment, but nevertheless are in general use, both as commercially available kits and in configurations developed in individual laboratories.
Fluorescence Polarization
Linear polarized light excites those fluorophores that are in line with the plane of polarization. Fluorescent molecules will emit light in this same plane if they are rotating very slowly or are stationary; if they are tumbling rapidly in solution, they emit light in different planes (i.e., are depolarized or scattered). If a fluorophore is conjugated to a large molecule (e.g., ubiquitin), its overall rotation is much slower than its rotation as a free molecule in solution, and the ratiometric measurement of the amounts of polarized and depolarized light, spectroscopically determined in high-throughput fashion, yields a quantitative estimate of the relative conjugation status of a fluorophore. A protease (e.g., a DUB), converts a conjugated fluorophore (highly polarized) to a free fluorophore (depolarized), so the average amount of polarization (expressed in millipolarization (mP) units) decreases as hydrolysis proceeds, and inhibition of hydrolysis reduces the decrease in polarization observed. The technique is minimally sensitive to fluorescent or quenching compounds and can be effectively used to screen for DUB inhibitors. Moreover, this type of assay can be designed in such a way that differences among individual DUBs can be exploited and that, in at least one recently reported study, isopeptide cleavage can be measured. 63
Fluorescence Resonance Energy Transfer
In this detection method, energy is transferred from one chromophore to another; the donor, in an excited state, transfers energy to the acceptor with an efficiency that is proportional to the sixth power of the distance between them, making FRET highly sensitive to small intermolecular distances. 64 Measurements of FRET efficiency can be used to determine if two fluorophores are within a certain distance of each other; 65 this property is the basis of FRET-based measurement of DUB-catalyzed cleavage of ubiquitin, which results in the separation of appropriate FRET donor–acceptor pairs.
Assays That Measure Isopeptide Bond Cleavage
In recent years, numerous assays characterized by increasing levels of sophistication imparted by advances in structural biology and biochemistry have been developed to measure the cleavage of Ub from its binding partner protein by DUB activity. Cleavage substrates are now available that use ubiquitin linked by an isopeptide bond to a fluorescent molecule (e.g., a peptide bearing a fluorescent tag); these substrates can be adapted to FP- or FRET-based assays 63 (see above), and they address the false positives, quenching issues, and linear peptide bond cleavage associated with the first DUB screening assays.
To mimic ubiquitin conjugated through an isopeptide bond to an ϵ-amino group of a lysine on its protein target, di-ubiquitin substrates have been developed, in which the carboxy terminus of a ubiquitin molecule is linked through an isopeptide bond to a lysine of a second ubiquitin molecule. The di-ubiquitin molecule becomes an internally quenched fluorescent pair if one ubiquitin is labeled with a single molecule of a fluorescent reporter molecule and the other with a highly efficient quenching fluorophore. Cleavage of the di-ubiquitin by DUBs leads to separation of the fluorophore from the quencher and subsequently to a fluorescence signal. Assays using this class of substrate measure cleavage of a true isopeptide linkage. In addition, it has been possible to construct substrates that contain various types of isopeptide linkage in terms of the ubiquitin lysine (e.g., K63, K48, and K11), and these substrates are being used in ongoing DUB drug-discovery efforts.66–69 Ubiquitylation does not impart a simple one- or two-dimensional code, and as more is learned about the kinds of ubiquitin chains associated with the physiology of disease-related DUBs (e.g., which lysines and what kinds of branching are involved), it may be advantageous to assay specific types of cleavage for translational and therapeutic purposes; 20 multi-ubiquitin substrates could enable such an undertaking.
Emerging Approaches for Discovering Improved DUB Effector Molecules
Given the somewhat disappointing results of past efforts to discover potent, selective, and tractable inhibitors of DUBs by traditional screening for molecules that affect the activity of the highly conserved catalytic site, newer approaches include protein-engineering strategies that take a more nuanced view of the active site as well as screening and design approaches that focus on noncatalytic sites.
Exploiting Ubiquitin Binding to the DUB Catalytic Core
In early 2013, two publications appeared that addressed the lack of potent, selective DUB inhibitors through novel, related protein-engineering approaches. The first, from Genentech, considered the rather large degree of conformational flexibility of the 7.6 kDa ubiquitin molecule as a hindrance to tight binding of ubiquitin to the DUB catalytic core (whose affinity for ubiquitin is greatly reduced compared to its affinity for the intact enzyme, as judged by KD).70–72 Using computational and phage-display methods, ubiquitin variants were isolated and tested for binding and biological properties; several were identified that bound the USP7 catalytic core and inhibited the purified DUB with nanomolar potency. One of these, expressed in cancer-derived cells, achieved proof of concept by inhibiting endogenous USP7 and upregulating its pharmacodynamic markers, p53 and p21. A second study, performed by a group at the University of Toronto, also used combinatorial libraries of ubiquitin variants. They developed highly potent inhibitors of four DUBs—USP21, USP8, USP2a, and OTUB1—and performed crystallographic studies on the DUB–variant complexes and functional studies with cultured cells. 73 The key to selectivity of the ubiquitin variants is the rather large contact surface between ubiquitin and the DUB, which is composed of residues that are not conserved; when the normally plastic ubiquitin molecule is forced into a particular conformation, it binds with high affinity to a discrete number of ubiquitin pathway enzymes at their catalytic sites. The achievement of predicted functional effects in cells by the correct variant supports the notion that these variant effector molecules act selectively. Although the practical application of this work is concerned primarily with translational or mechanistic studies, it is conceivable that peptidomimetic molecules could be designed and developed as potent and selective DUB inhibitors.
Exploiting Binding Sites outside the Catalytic Core
A drug that works by inhibiting or activating a DUB does not necessarily have to be a competitive inhibitor of ubiquitin binding in the catalytic pocket. It is possible that inhibitors with improved selectivity and other desirable properties could be discovered if screening and drug design are focused on a relatively weakly conserved portion of the target DUB molecule. Investigators have begun to use this strategy for the discovery and design of inhibitors of various cysteine proteases. Hagel and colleagues found irreversible inhibitors targeting noncatalytic cysteines in a structurally unique portion of HCV protease, 74 a strategy that can be adapted to the discovery of effectors of numerous therapeutically relevant DUBs. Nonactive site inhibitors can be the goal of HTS by the use of biophysical methods, as described below, in addition to rational design and virtual screening.
Assessment of Ligand–Protein Binding in Biophysical Assays
Alternative approaches to a biochemical endpoint (deubiquitylation) assay model include various biophysical assays. As one example, thermal shift technology 75 can be used to monitor the change in the conformational stability of a protein (e.g., a DUB) when it binds a high-affinity ligand. Binding stabilizes the protein and increases its melting temperature. Compounds identified as DUB binders in such an assay can be characterized further in secondary assays for biochemical activity; alternatively, to preclude or discriminate highly conserved, nonselective “catalytic-site” binders, screening can be performed using DUBs that either lack the catalytic core or contain an altered core. The thermal shift method uses differential scanning fluorometry to measure the shift in the melting curve of a protein when it is bound by a ligand (e.g., a small molecule) and stabilized. The methodology is adaptable for moderate-throughput screening using proteins or protein fragments, and it has been used to screen compounds in primary drug-discovery screens in the pharmaceutical industry, including screens with ubiquitin E3 ligases.75–77
Another biophysical method that has been used to identify molecules that bind enzymes is surface plasmon resonance (SPR). 78 SPR detects changes in the refractive index of a solvent at a sensor surface and can be used to identify protein binders. 79 Although SPR is typically a low-throughput method, advances in instrumentation are increasing its throughput. At present, SPR technology can accommodate moderate-throughput operations (~5000 fragments). The technique was used to identify compounds that bind the ubiquitin E3 ligase parkin; notably, new scaffolds and leads were identified that differed from compounds discovered in earlier enzyme activity-based screening and were subsequently abandoned owing to their inability to improve potency. 78 This technology is readily applicable to other enzymes, including DUBs. Thus, data emerging from the use of thermal shift and SPR screening suggest that high-quality, tractable molecules acting on ubiquitin pathway enzymes can be discovered, and it is likely that high-throughput biophysical methods such as these will be used extensively not only to characterize or confirm binding of effector molecules to their targets, but also as screening modalities.
Conclusions
DUBs continue to be an attractive therapeutic target class, because new roles for these enzymes in various diseases are uncovered each year and target validation follows closely behind. After a decade of HTS and chemical optimization of hits, several selective catalytic-site DUB inhibitors have been described, but to date none of these compounds have entered late-phase preclinical testing. As a consequence, discovery efforts that target DUBs are currently lagging, relative to programs aimed at the proteasome or E3 ligases. Recent work with ubiquitin mutants demonstrates that it is possible to inhibit individual DUBs with impressive potency and selectivity. Biophysical assays have also shown promise when adapted to DUBs and E3 ligases; they may succeed in identifying small molecules that bind outside the active site to inhibit DUB activity selectively. Active site inhibitors discovered with the use of better-discriminating screening modalities such as substrate-specific activity assay formats, FP or FRET assays that selectively measure particular Ub–protein configurations, or targets with engineered active sites, may also offer potential in the continuing search for DUB inhibitors for the clinic.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.