
Abstract
The ubiquitin-proteasome system is central to the regulation of numerous cellular events, and dysregulation may lead to disease pathogenesis. E3 ubiquitin ligases typically function in concert with E1 and E2 enzymes to recruit specific substrates, thereby coordinating their ubiquitylation and subsequent proteasomal degradation or cellular activity. E3 ligases have been implicated in a wide range of pathologies, and monitoring their activity in a rapid and cost-effective manner would be advantageous in drug discovery. The relative lack of high-throughput screening (HTS)–compliant E3 ligase assays has significantly hindered the discovery of E3 inhibitors. Herein, the authors describe a novel HTS-compliant E3 ligase assay platform that takes advantage of a ubiquitin binding domain’s inherent affinity for polyubiquitin chains, permitting the analysis of ubiquitin chain formation in an E3 ligase-dependent manner. This assay has been used successfully with members of both the RING and HECT families, demonstrating the platform’s broad utility for analyzing a wide range of E3 ligases. The utility of the assay platform is demonstrated by the identification of inhibitors of the E3 ligase CARP2. As the number of E3 ligases associated with various disease states increases, the ability to quantitate the activity of these enzymes in an expeditious manner becomes imperative in drug discovery.
Introduction
P
Ubiquitin is covalently attached to proteins by the coordinated action of 3 enzymes—an E1 activating enzyme, an E2 conjugating enzyme, and an E3 ligase. The mechanistic aspects of ubiquitin conjugation have been elucidated and are now being exploited for drug discovery. These enzymes function in sequential steps, each of which can be seen as a potential therapeutic target. 1,6 The E3 ubiquitin ligases function in concert with E1 and E2 enzymes to recruit specific substrates, thereby coordinating their ubiquitylation for subsequent proteasomal degradation and or translocation. 4 The approval of the proteasome inhibitor bortezomib (Velcade®) for the treatment of multiple myeloma clearly validates the ubiquitin proteasome pathway as a source of new drugs. Bortezomib is an active drug, but its utility is limited by toxic side effects. 7 The targeting of enzymes and regulatory pathways upstream of the proteasome, on the other hand, would affect a small population of target proteins and thus offer selective therapies of target-linked diseases. For that reason, E3 ligases, an enzymatic family consisting of ~600 proteins, represent an attractive drug target because of their limited number of substrates and selective regulatory pathways. E3 ubiquitin ligases are the final enzymes in the ubiquitin conjugation cascade, bestowing substrate specificity. For these reasons, various pharmaceutical companies and academic laboratories have sought to identify novel, specific E3 inhibitors. However, results have been disappointing, considering that to our knowledge, only a single E3 inhibitor has entered clinical trials to date (Johnson & Johnson, JNJ-26854165, acts against HDM2). Several different types of assays have been developed for E3 inhibitor screening, but obstacles dealing with the complexity of the E3 ligase family have impeded drug discovery in this area. 8 As the number of E3 ligases associated with various disease states increases, 9-12 the ability to test these enzymes in an expeditious manner becomes very important. The lack of mechanistically relevant, cost-effective E3 ligase assays amenable to high-throughput screening (HTS) has significantly hindered the discovery of E3 inhibitors.
Polyubiquitin binding domains are structural motifs found in proteins; they are employed to recognize ubiquitin moieties conjugated to substrates in the cell. These ubiquitin binding domain–containing proteins can play a role in protein degradation as well as in nonproteasomal cellular processes. For instance, specific subunits of the proteasome 19S cap can serve as ubiquitin receptors, capable of recognizing ubiquitin-conjugated proteins for degradation. 13 For example, S5a from higher eukaryotes and Rpn13 are viewed as the major contributors for proteasome ubiquitin recognition. 14 Many polyubiquitin binding domains have been identified and characterized in a wide variety of proteins, and they are divergent in sequence (20-150 residues), structure, and patterns of ubiquitin recognition. 15-17 Binding between these domains and monoubiquitin has been reported to be of low or moderate affinity; however, the binding affinity is strongly enhanced by the recruitment of multiple ubiquitin moieties in the form of polyubiquitin chains. 17
Herein, we describe a novel E3 ligase assay platform that takes advantage of a ubiquitin binding domain’s inherent affinity for ubiquitin, 18 preferentially binding polyubiquitin relative to monoubiquitin and permitting the analysis of chain formation in an E3 ligase-dependent manner. Specifically, the immobilization of UBA domains allows for the capture and isolation of polyubiquitylated proteins from in vitro ubiquitylation reactions. Subsequently, the relative amounts of polyubiquitylated proteins are measured in an enzyme-linked immunosorbent assay (ELISA) format, enabling the quantification of E3 ligase activity. This assay platform has been used successfully with members of both the RING and HECT families, demonstrating the platform’s broad utility for analyzing a wide range of E3 ligases. In addition, using the E3 ubiquitin ligase CARP2 as an example, we demonstrate that this assay can be configured for HTS. Our screening campaign targeted CARP2, owing to the role that it plays in the ubiquitylation of caspase-8 and -10. 19 Caspases are cysteine proteases that destroy cellular targets during apoptosis and are negatively regulated by inhibitors of apoptosis proteins (IAPs), but caspase-8 and -10 are refractory to IAPs and have found to be negatively regulated by the CARP family. Inhibition of expression of the CARP family in tumor cell lines results in suppression of cancer cell growth, highlighting the importance of this family in drug discovery. 19
Materials and Methods
Unless stated otherwise, all reagents were obtained from Sigma (St. Louis, MO) and were at a minimum of reagent grade or better. Plasmids encoding Ube1, MuRF1, and Hrd1 cDNAs were purchased from Open Biosystems (Huntsville, AL), whereas other vectors were generous gifts (please see Acknowledgments).
Cloning and protein expression
The cloning, expression, and purification from BL21 (DE3) bacteria of the SUMO-UBA2 from Rad23 (residues 351-398), K48 only (K6R, K11R, K27R, K29R, K33R, and K63R) ubiquitin, 6His-Ube1, 6His-UbcH5b, 6His-UbcH5c, 6His-UbcH7, SUMO-CARP2, SUMO-CARP2 (H333A), SUMO-MuRF1, 6His-E6AP, and SUMO-Hrd1 (residues 235-617) were performed using standard molecular biology techniques,20 using SUMOpro fusion vectors (LifeSensors, Inc., Malvern, PA) where denoted. All constructs were fully sequenced to verify wild-type sequence or to check introduced point mutations.
UBA assay for E3 ligase autoubiquitylation
MicroFluor 96-well medium binding plates (Thermo Fisher Scientific, Rockford, IL) were preincubated with purified SUMO-UBA2 before washing and blocking with 3%(w/v) bovine serum albumin (BSA). SUMO-UBA2 is immobilized nonspecifically through hydrophobic interactions by passive adsorption. In a typical assay, varying concentrations of an E3 ligase were incubated with 5 nM Ube1, 100 nM E2, and 1.1 µM K48 Ub in a 50-µL reaction buffer (50 mM Tris-HCl [pH 8.0], 5 mM MgCl2, 0.2 mM adenosine triphosphate [ATP], and 1 mM β-mercaptoethanol) in each well. After incubation at room temperature for a fixed period of time within the initial linear range, the plates are washed 3× with phosphate-buffered saline with 0.1% (v/v) Tween (PBST) with a platewasher (BioTek, Winooski, VT). E3 autoubiquitylation was detected by sequential 1-h incubations with rabbit anti-ubiquitin (1:50 dilution) (Sigma) and antirabbit–fluorescein isothiocyanate (FITC) or antirabbit–horseradish peroxidase (HRP) (1:10,000; Jackson ImmunoResearch Laboratories, West Grove, PA) in 5% (w/v) BSA/PBST; the plates were washed with PBST after each incubation. All the bulk reagent dispensing was performed by a microflo broadcast dispenser (BioTek). Relative levels of ubiquitylated product were determined by a luminescence plate reader (EnVision, PerkinElmer, Waltham, MA) using an enhanced chemiluminescence (ECL) reagent (Millipore, Billerica, MA). Similar experiments were conducted for the E1 and E2 optimization, where serial dilutions of E1 or E2 enzymes were performed to detect signal saturating concentrations, ensuring non-rate-limiting conditions. This technology platform is distributed by LifeSensors, Inc (www.lifesensors.com).
Ubiquitin chain recognition assay
Monomeric ubiquitin or purified ubiquitin chains 2-7 (Boston Biochem, Cambridge, MA) were incubated in 50 µL reaction buffer in precoated and blocked SUMO-UBA2-coated 96-well plates. After incubation at room temperature for 30 min, the plates were washed 3× with PBST with a platewasher. Ubiquitin chain recognition was detected as described above.
Gel-based in vitro autoubiquitylation assay
Dose ranges of SUMO-CARP2 were incubated in reaction buffer, with 5 nM Ube1, 100 nM UbcH5c at room temperature for 20 min. Subsequently, the reactions were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) under reducing/denaturing conditions. Following transfer to nitrocellulose, the blot was placed in boiling water for 5 min before blocking with 5% (w/v) nonfat dried milk/PBST and probing sequentially with anti-ubiquitin (Sigma) and antirabbit-HRP (Jackson ImmunoResearch Laboratories) before visualizing with an ECL reagent (Pierce, Rockford, IL) using a Fuji digital imager.
Apparent Km determination
UBA2 precoated, preblocked 96-well plates were prepared as described above, and serial dilutions of E1 activating enzyme or E2 conjugating enzyme were added to ubiquitin (1 µM final) and E6AP (16 nM final) in the 96-well plate before initiating E3 autoubiquitylation by the addition of ATP to a final concentration of 0.2 mM in 50 mM Tris-HCl (pH 8.0), 5 mM MgCl2, and 1 mM β-mercaptoethanol. The UbcH5c concentration was 100 nM when determining Ube1 saturation. The concentration of Ube1 was 5 nM when determining E2 affinity. After 1 h at room temperature, the reaction plate was washed 3× with PBST, and the presence of polyubiquitin was detected as described above. The relative velocity (RLU/min) was fit to the Michaelis-Menten equation using Prism software (GraphPad, San Diego, CA) to determine apparent Km values. As this coupled assay does not report absolute velocities, it is not possible to determine Vmax values using this assay platform.
HTS
From our internal diversity-based collection of small molecules, 10-mM test compounds were diluted in 100% DMSO to a concentration of 2.5 mM. Then, 96-well plates were precoated with UBA2 and blocked as described above. Subsequently, 0.5 µL of 2.5 mM test compound was preincubated at a concentration of 50 µM with 25 µL of 10 nM Ube1, 200 nM UbcH5c, and 16 nM of CARP2 in 50 mM Tris-HCl (pH 8.0), 5 mM MgCl2, and 1 mM β-mercaptoethanol for 30 min. The autoubiquitylation reaction was initiated by the addition of 25 µL of 1.1 µM ubiquitin and 0.4 mM ATP in 50 mM Tris-HCl (pH 8.0), 5 mM MgCl2, and 1 mM β-mercaptoethanol. After 20 min, the reactions were terminated by washing as described above. Ubiquitylated products were detected as described above. Each assay plate was internally controlled using final concentrations of 10 mM N-ethyl-maleimide (NEM) and 1% (v/v) DMSO for 100% and 0% inhibition, respectively. Percent inhibition was calculated using the following equation: [1 – ((RLUCompoundl – RLUNEM)/(RLUDMSO – RLUNEM))]*100. The mean percent inhibition of the test compounds was 14.9%, and the standard deviation was 11.3%. Hits were identified as compounds that inhibited CARP2 autoubiquitylation by greater than 4 standard deviations from the mean percentage inhibition of the screen. Initial hit confirmation was performed by rescreening hits from the primary screen as described above.
Results
Immobilized ubiquitin binding domain captures ubiquitin chains
The Saccharomyces cerevisiae Rad23 and its human homologue hHR23A contain 2 ubiquitin binding domains, UBA1 and UBA2, that collectively contribute to the binding of poly- ubiquitin chains in the cell. Individually, as truncated domains, these motifs can fold into stable, independent structures that can bind polyubiquitin chains autonomously. 18 In particular, the ubiquitin binding domain UBA2 from Rad23 has been reported to preferentially recognize lysine 48-linked tetra-ubiquitin chains. 18,21
Initially, we determined if we could specifically bind polyubiquitin conjugates using immobilized UBA2. Earlier studies had demonstrated that recognition of longer ubiquitin chains by immobilized individual UBA domains was more efficient when linked to the fusion protein glutathione S-transferase (GST).
22
In our studies, we recombinantly expressed UBA2 from Rad23 linked to SUMO and immobilized it in 96-well microplates to analyze the domains’ affinity for ubiquitin when incubated with either monoubiquitin or purified K48 (Ub2-7) ubiquitin chains. After incubation, multiple wash steps with phosphate-buffered saline (PBS) were conducted before probing the plate with an anti-ubiquitin antibody, followed by an antirabbit-FITC secondary antibody). Data in
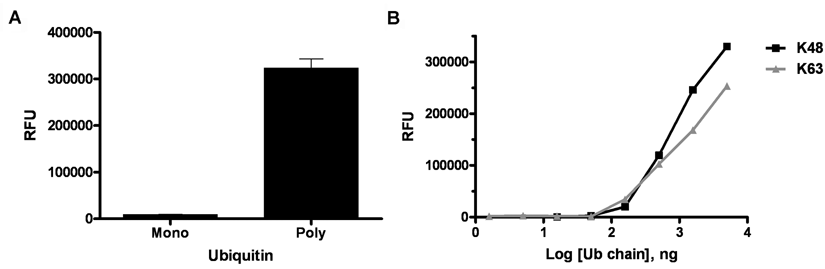
UBA2-coated plates preferentially bind polyubiquitin. (
To analyze the affinity of UBA2-coated plates for specific ubiquitin chain linkages, K48 and K63 ubiquitin chains were evaluated. These 2 types of ubiquitin chain are prevalent in the cell and function as components of the proteolytic and nonproteolytic signaling pathways for E3 ligase targeted substrates, respectively. As stated above, UBA2 has been reported to preferentially bind K48-linked tetra-ubiquitin
18
; however, in the absence of K48-linked chains, ubiquitin binding domains retain the ability to bind K63-linked chains, albeit with a lower affinity.
18
For our evaluation, serial dilutions of purified K48 and K63 (Ub2-7) polyubiquitin chains were incubated in UBA2-precoated plates. Interestingly, in this assay format, both lysine chain linkages exhibited similar dose-dependent binding (
Detection of E3 ligase autoubiquitylation activity
Given that UBA-coated plates can exclusively bind polyubiquitin chains of at least 2 different linkages, we determined the feasibility of using the UBA2-coated plates in a sandwich ELISA analyzing E3 ligase activity (
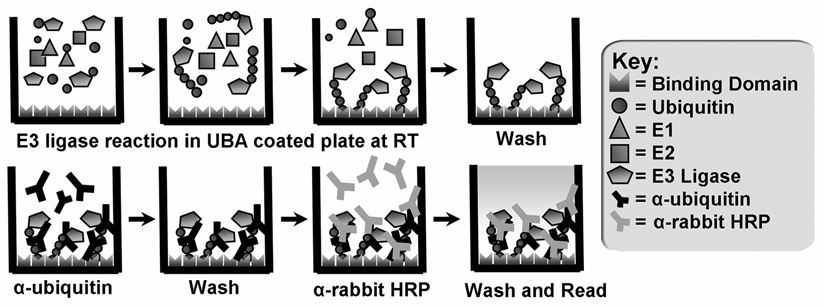
Schematic model of the E3 ligase assay platform. Ubiquitylation reaction enzymes are incubated at room temperature (RT) in a UBA2-coated 96-well microplate before washing to remove the enzyme components and any unbound ubiquitin. The polyubiquitylated product adheres to the plate because of the ubiquitin binding domain’s affinity for polyubiquitin chains. Chains are quantitated in a luminescence plate reader after the sequential use of anti-ubiquitin and anti-rabbit horseradish peroxidase (HRP) antibodies to monitor E3 ligase-dependent ubiquitylation.
Initially, we studied the RING E3 ligase CARP2 (caspase-associated RING finger protein), an enzyme associated with caspase degradation and apoptosis.
19,23
To increase the dynamic range, we replaced the fluorescence readout with a luminescence readout. This adjustment led to an increased dynamic range due to the ECL amplification reaction (data not shown). As expected, CARP2 autoubiquitylation was observed only when Ube1 (E1), UbcH5c (E2), CARP2, and ubiquitin were present (
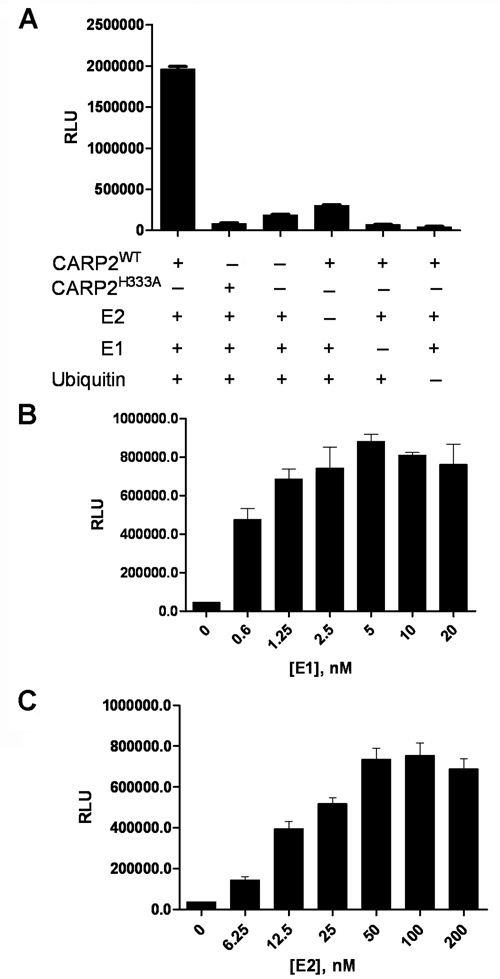
The enzyme-linked immunosorbent assay (ELISA) is E3 dependent. (
To develop an assay for identifying modulators of E3 ligases, ubiquitylation reactions should be E3 dependent. To ensure that a ubiquitylation reaction was limited by CARP2, experiments were conducted to identify saturating E1 and E2 concentrations. By determining CARP2 activity in the presence of excess UbcH5c and variable concentrations of Ube1, we were able to determine a saturating concentration of Ube1 at 5 nM for the reaction (
The current gold standard for measuring E3 activity is time-consuming gel-based analysis of autoubiquitylation reactions. We sought to directly compare the sensitivity of the ELISA platform with gel-based analysis by testing dose ranges of CARP2 in the 2 assay formats. Data presented in
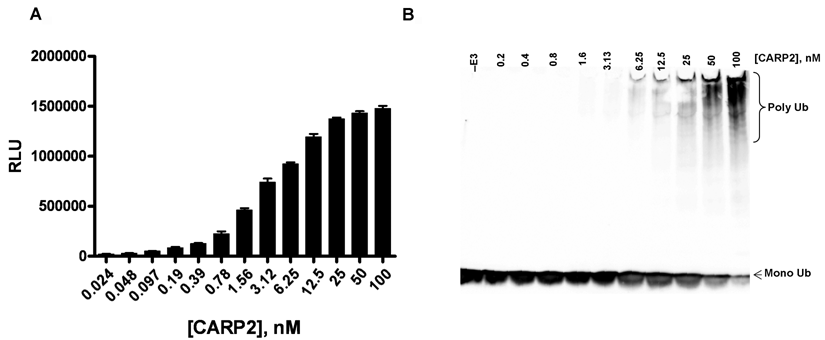
(
The ELISA format is functional for a broad spectrum of E3 ligases
To confirm the utility of this assay platform to measure the activities of multiple E3 Ub ligases, we tested RING (MuRF1 and Hrd1) and HECT domain (E6AP) E3 ligases with the promiscuous E2 enzyme, UbcH5c.
24
Data from these studies illustrated that the ELISA platform had the utility to measure both RING and HECT E3 ligase autoubiquitylation in a dose-dependent manner (
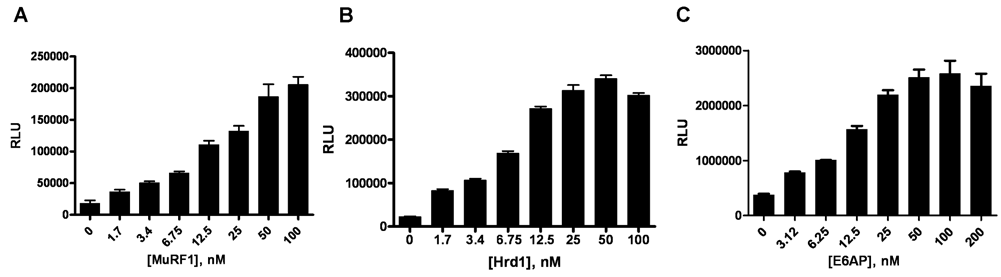
The enzyme-linked immunosorbent assay (ELISA) format is capable of reporting activity for both RING and HECT E3 ligases. (
It is increasingly becoming recognized that the identification of the cognate E2 for an E3 is an important step in developing a valid assay for measuring E3 ligase activity. In many cases, the cognate E2 is not known. For example, the HECT E3 ligase E6AP previously has been reported to exhibit activity with at least 3 conjugating enzymes—UbcH5b, UbcH5c, and UbcH7.
25
One way to determine the appropriate E2 for an E3 is to measure the Km value of individual E2s for an E3. The Km encompasses the affinity of a substrate for an enzyme. By comparing the Km values, the optimal E2 partner can be determined. We sought to use the ELISA to determine the optimal conjugating enzyme for E6AP by probing the affinity of UbcH5b, UbcH5c, and UbcH7 for E6AP (
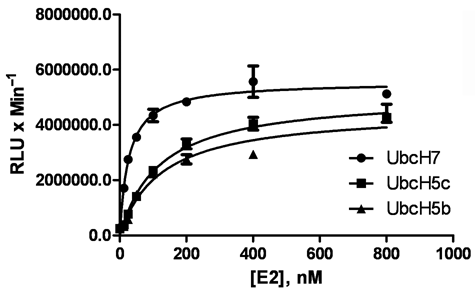
UbcH7 is the appropriate E2 enzyme for E6AP. Serial dilutions of UbcH7, UbcH5b, and UbcH5c were added to E1 activating enzyme, E6AP, and ubiquitin. After the addition of adenosine triphosphate (ATP), E6AP-dependent ubiquitylation was monitored and reported using the UBA-coated plate. The data were fit using Prism software to determine apparent Km values. Data are mean ± SD of a representative experiment from 3 independent experiments.
The ELISA platform is configurable for HTS
Determining E3 ligase activity enables the discovery and characterization of novel modulators for E3 Ub ligase activity. We investigated the ability of the ELISA platform to measure CARP2 activity in an HTS format. Our screening campaign targeted CARP2, an E3 ligase that ubiquitylates caspase-8 and -10.
19
In our assay format, CARP2 was screened at a final concentration of 8 nM. This final concentration was chosen in view of the fact that the effective dose (ED50) for CARP2 was approximately 4 nM, the dose that generates half of the maximal signal (data not shown). CARP2 autoubiquitylation exhibited linear kinetics over the first 30 min. Initially, we determined that the assay could tolerate DMSO concentrations ≤5% with negligible loss of assay performance (data not shown). A commonly accepted parameter of the adaptability of an assay to an HTS format is the Z′ value, which takes into account the dynamic range and assay variability. Specifically, a Z′ value of ≥0.5 represents an assay that is robust and reproducible in an HTS format.
26
As no inhibitors of CARP2 are available, we used the sulfhydryl alkylating agent NEM, which is a known inhibitor of E1 and E2 activities to determine the CARP2 ELISA Z′ in a 96-well plate format. Data from these experiments revealed that the CARP2 assay had a Z′ of 0.76 ± 0.05 (
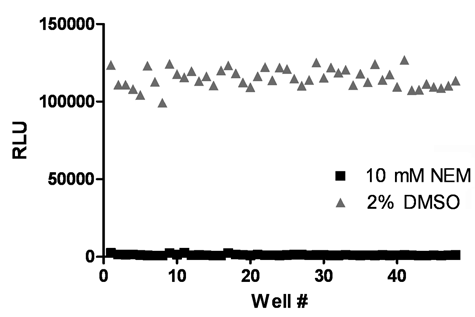
The CARP2 enzyme-linked immunosorbent assay (ELISA) is suitable for high-throughput screening (HTS). CARP2 was validated for HTS in 96-well plates with a Z′ of 0.76 ± 0.05 from 3 independent experiments. Data shown from a representative experiment.
Identification of E3 ligase inhibitors using the ELISA platform
To further demonstrate the utility of the ELISA platform for HTS, we screened a small-molecule diversity library consisting of ~12,700 compounds for inhibitors of CARP2 autoubiquitylation. The assay performed consistently and remained within acceptable parameters over the course of the screen (assay Z′ 0.64 ± 0.08). We identified 71 hits (0.56% hit rate) from the primary screen, where a hit was defined as a compound inhibiting CARP2 autoubiquitylation activity by greater than 4 standard deviations from the mean percentage inhibition of the screen (
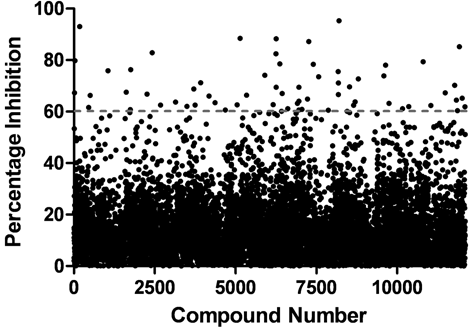
Identification of CARP2 inhibitors. CARP2 was screened against a small-molecule diversity-based library, and hits were identified as compounds that inhibited CARP2 autoubiquitylation activity by greater than 4 standard deviations from the mean percentage inhibition (indicated by the dotted line).
Discussion
There is an intense interest in the pharmaceutical and academic community to identify novel modulators of ubiquitin E3 ligase activity. To aid in the discovery process, a number of different E3 ligase activity assays have been described.
27-30
However, these assays have inherent limitations that constitute liabilities when screening with purified enzymes, for example, requirements for the immobilization or fluorescence tagging of critical components such as E3 ligases/substrates involved in ubiquitylation. Likely as a consequence of these limitations, only 1 E3 inhibitor to our knowledge has entered clinical trials. Here, we describe the generation and implementation of a novel assay platform for E3 ligases that uses ubiquitin binding domains to bind assembled polyubiquitin chains for detection. This assay circumvents some of the limitations that apply to currently used E3 ligase assay systems. This novel assay allows all of the components involved in ubiquitylation to react freely in solution in their native states, before E3-dependent chain formation, capture, and subsequent detection. Importantly, when the ELISA format was directly compared to gel-based detection, the ELISA format was shown to be a more sensitive method for detecting autoubiquitylation (
Initial experiments demonstrated that the ubiquitin binding domain UBA2 immobilized on a microplate was able to bind polyubiquitin chains selectively with respect to monoubiquitin and did not discriminate between the 2 commonly formed ubiquitin chain linkages in the cell, K48 and K63 (
Our ligase activity platform is configured as an ELISA with a luminescence readout, providing a simple and efficient method to screen for inhibitors of E3 ligase activity. Several features make it a desirable approach, including high sensitivity and the removal of test compounds prior to assay readout, thus obviating any interference in the assay readout. Initially, we configured the assay for screening in 96-well plates and confirmed that the assay was suitable for screening (Z′ = 0.76 ± 0.05;
The data presented within this study demonstrate that our novel UBA-based assay platform can be used as a robust and sensitive E3 ligase reporter system. Given the intense interest in the field to identify clinical candidate modulators of E3 ligase activities, there has been poor progress on candidates advancing to the clinic. We believe one reason for this disappointing metric is the currently employed E3 Ub ligase assay systems. As E3 ligases are an emerging therapeutic target for a variety of diseases, the E3 Ub ligase ELISA described here will be an improved tool to identify novel modulators of targeted ligases.
Footnotes
Acknowledgements
We sincerely thank Drs. Kiran Madura, Wafik El-Deiry, Arthur Haas, Nafar Nawaz, Serge Fuchs, and Jim Strickler for their help and contribution.This work was supported by National Cancer Institute (NCI) and National Institutes of Health (NIH) grants to Progenra, Inc. (CA120227 and CA128191).