
Abstract
Alterations in synaptic transmission have been implicated in a number of psychiatric and neurological disorders. The discovery of small-molecule modulators of proteins that regulate neurotransmission represents a novel therapeutic strategy for these diseases. However, high-throughput screening (HTS) approaches in primary neurons have been limited by challenges in preparing and applying primary neuronal cultures under conditions required for generating sufficiently robust and sensitive HTS assays. Synapsin I is an abundant presynaptic protein that plays a critical role in neurotransmission through tethering synaptic vesicles to the actin cytoskeleton. It has several phosphorylation sites that regulate its modulation of synaptic vesicle trafficking and, therefore, the efficacy of synaptic transmission. Here, we describe the development of a rapid, sensitive, and homogeneous assay to detect phospho-synapsin I (pSYN1) in primary cortical neurons in 384-well plates using AlphaScreen technology. From results of a pilot screening campaign, we show that the assay can identify compounds that modulate synapsin I phosphorylation via multiple signaling pathways. The implementation of the AlphaScreen pSYN1 assay and future development of additional primary neuronal HTS assays provides an attractive approach for discovery of novel classes of therapeutic candidates for a variety of CNS disorders.
Introduction
Synaptic transmission is the fundamental basis of neuronal communication in the brain. 1 During this process, action potential–mediated calcium influx causes the exocytosis of synaptic vesicles from the presynaptic terminal and release of neurotransmitter into the synaptic cleft to activate postsynaptic receptors. 2 Efficient synaptic vesicle mobilization and recycling are required to maintain the releasable pool of synaptic vesicles, which is essential for synapses to function over a wide range of activity levels. 3 Alterations in synaptic transmission are known to be involved in a number of CNS disorders,4 –8 including autism, epilepsy, Alzheimer disease, schizophrenia, and Huntington disease, suggesting that the identification of compounds that can restore normal synaptic function could provide the basis for discovery of novel classes of therapies targeting these diseases.
Synapsin I, a member of a family of neuron-specific phosphoproteins, regulates synaptic vesicle release and recycling by modulating the mobilization of synaptic vesicles during neurotransmission. 9 Synapsin I can be phosphorylated at eight residues, termed sites 1 to 8, by the following kinases: cAMP-dependent protein kinase A (PKA) at site 1, calcium-calmodulin–dependent kinase II (CaMKII) at sites 2 and 3, mitogen-activated protein kinase (MAPK) at sites 4 and 5, cyclin-dependent kinases 1 and 5 (CDK1/5) at sites 6 and 7, and Src kinase at site 8. 10 These sites are dephosphorylated by protein phosphatase 2A (sites 1–3) and calcineurin (sites 4–7).10,11 The functions of synapsin I are tightly regulated by the phosphorylation status of these residues. Unphosphorylated synapsin I tethers synaptic vesicles in the reserve pool through its association with actin, microtubules, and neurofilaments. 12 Phosphorylation of synapsin I on sites 1 to 7 decreases its affinity for cytoskeletal components, 13 resulting in mobilization of synaptic vesicles for release. 14 In addition, activity-dependent dephosphorylation of site 6 by calcineurin is required for vesicle turnover during high-frequency synaptic activity. 15
Alterations in the phosphorylation of synapsin I may play a role in diseases associated with aberrant synaptic transmission. Indeed, reduced phosphorylation at site 1 has been observed in Alzheimer disease postmortem tissues, 16 and hyper- and hypophosphorylation at sites 3 to 5 and 6, respectively, have been observed in a Huntington disease mouse model. 17 The important role of synapsin I in synaptic transmission and its significance in CNS disease biology make it an attractive target for the identification of small molecular modulators of synaptic function. Such a screening approach would ideally be undertaken using primary neuronal cells, which contain the relevant signaling pathways for the regulation of synapsin I phosphorylation status. However, despite the therapeutic potential of such screening strategies, compound screening efforts in primary neurons have been limited due to challenges in generating robust and uniform cultures for high-throughput screening (HTS) applications. In addition, most methods for measuring protein phosphorylation status rely on laborious techniques that are not amenable to HTS, such as Western blotting, enzyme-linked immunosorbent assay (ELISA), and radioimmunoassay.
Here, we describe a method for preparing primary neuronal cultures in 384-well plates and the development and validation of an HTS assay for detecting phospho-S549 (site 6) of synapsin I (pSYN1) in these cultures using AlphaScreen, a proximity-based, homogeneous immunoassay. 18 Using the AlphaScreen pSYN1 assay, we performed a pilot screen of ~10,000 compounds and identified a number of compounds that induce the dephosphorylation of synapsin I. Our results demonstrate that the AlphaScreen pSYN1 assay is a robust assay that can be implemented in HTS campaigns to identify synapsin I modulators.
Materials and Methods
Cell Culture
Primary cortical neuronal cultures were prepared from E18 Sprague-Dawley rat embryos. Fetal brains were isolated and placed in Petri dishes containing cold Hank’s buffered salt solution (HBSS) (Invitrogen, Carlsbad, CA). Cortices were extracted, and the meninges were removed. The cortices were placed in a new Petri dish with HBSS, cut into 1-mm pieces, and transferred to a 15-mL conical tube. Residual HBSS was aspirated, and the tissue was incubated in digest solution consisting of 2.5% trypsin (Invitrogen) and 1% DNAse (Sigma, St. Louis, MO) in HBSS at 37 ° C for 15 min. The tissue was washed twice with HBSS and resuspended in dissociation solution consisting of 1% DNAse in HBSS. A homogeneous suspension was generated by trituration using fire-polished Pasteur pipettes. The cell suspension was centrifuged at 1000 rpm for 10 min at 4 °C, and the pellet was resuspended in culture medium consisting of Neurobasal (Invitrogen), 2% B27 supplement (Invitrogen), 0.5 mM glutamine (Invitrogen), and 6 µM glutamate (Sigma). Cells were seeded using a Wellmate microplate dispenser (Matrix Technologies, Maumee, OH) at a density of 2 × 104 cells per well in 384-well poly-D-lysine–coated plates (BD Biosciences, Franklin Lakes, NJ) in a total volume of 75 µL per well. An additional 25 µL of medium was added to corner wells to compensate for potential volume loss from increased evaporation in these wells. Black/clear and white/opaque plates were used for immunofluorescence and AlphaScreen experiments, respectively. For 6-well cultures, cells were seeded at a density of 1 × 106 cells per well in 6-well poly-D-lysine–coated plates (BD Biosciences). The cell plates were incubated at 37 °C in a humidified 5% CO2 incubator for 20 days. Approximately 10 µL volume loss from evaporation per well was observed after 20 days for 384-well cultures. Medium was not changed or replenished during incubation to avoid physical perturbation of the cells during synapse maturation.
Western Blot Analysis of pSYN1
Primary cortical neuronal cultures were treated with 1% DMSO or compound for 1 h at 37 °C and then lysed with SureFire lysis buffer (PerkinElmer, Waltham, MA). Lysates were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and the separated proteins were transferred onto nitrocellulose membranes. The membranes were blocked for 1 h at room temperature with 10% instant nonfat milk in Tris-buffered saline (TBS) (100 mM Tris [pH 7.4], 150 mM NaCl). The blots were incubated with either anti-pSYN1 (anti–phospho(S549)-synapsin I [Novus, St. Louis, MO]) or anti–total synapsin I (Synaptic Systems, Göttingen, Germany) antibody diluted 1:2000 in 5% nonfat milk in TBST (TBS with 0.1% Tween 20) overnight at 4 °C with gentle shaking. After three 15-min rinses with TBST, the membranes were incubated with either anti–rabbit immunoglobulin G (IgG) or anti–mouse-IgG–horseradish peroxidase (HRP) antibodies (1:5000; Santa Cruz Biotechnology, Santa Cruz, CA) in 5% nonfat milk powder in TBST for 1 h at room temperature with gentle shaking. After three 15-min washes with TBS, the blots were developed with Visualizer Spray & Glow ECL Western Blotting Detection System (Millipore, Billerica, MA) and quantified using a UVP BioChemi System (UVP, Upland, CA).
Immunofluorescence of pSYN1 in Primary Neurons Cultured in 384-Well Format
The culture medium was aspirated using an ELx405 Select microplate washer (BioTek, Winooski, VT), and cells were fixed with 4% formaldehyde for 20 min and permeabilized with 0.3% Triton X-100 for 20 min. After washing with phosphate-buffered saline (PBS), the cells were treated with blocking buffer containing 0.1% Triton X-100 and 10% goat serum (Sigma) for 1 h at room temperature. The cells were incubated with anti-pSYN1 (1:1000) and anti-MAP2 (1:2000; Sigma) antibodies in blocking buffer overnight at 4 °C with gentle shaking. After washing with PBS, the cells were incubated with Alexa Fluor 555 goat anti–rabbit IgG (Invitrogen) and Alexa Fluor 488 goat anti–mouse IgG (Invitrogen) antibodies diluted 1:1000 in blocking buffer with 5% goat serum for 1 h at room temperature. Hoechst stain (1:10; Sigma) was included in the antibody mixture. After washing with PBS, the cells were visualized by microscopy (Axio Obzerver.Z1; Carl Zeiss MicroImaging LLC, Thornwood, NY) using a 40× objective.
Pilot Screen for Modulators of Synapsin I Dephosphorylation
Medium plates containing Neurobasal medium (24 µL/well) were freshly prepared. Compound plates were made in 384-well polypropylene plates (Greiner Bio-One, Monroe, NC). Column 23 contained 100% DMSO in each well. The positive control, ionomycin (Sigma), was prepared in 100% DMSO and added as a single-point concentration to each well in column 24. The final ionomycin concentration in the assay was 10 µM. Using a JANUS automated workstation (PerkinElmer), contents of each medium plate were aspirated, dispensed into a compound plate, and mixed 10 times. The mixture was then dispensed into a primary neuronal culture plate above the cell monolayer and gently mixed twice. The cell plate was incubated at 37 °C in a humidified 5% CO2 incubator for 1 h. After compound treatment, the AlphaScreen pSYN1 assay was performed. A pilot library containing ~10,000 compounds representing a subset of both our diverse and focused small-molecule libraries was screened in a single point at 10 µM. Hits were validated by cherry-picking in duplicate and tested to eliminate false positives using the AlphaScreen TruHits (PerkinElmer) kit. Concentration-response studies were performed in quadruplicate.
AlphaScreen pSYN1 Assay
All reagents were supplied in a custom-developed AlphaScreen kit (PerkinElmer). After compound treatment, the compound-medium mixture was aspirated, and cells were lysed with 5 µL of lysis buffer supplemented with protease inhibitor cocktail (Sigma). A mixture of biotinylated anti–total synapsin I antibody (Synaptic Systems) and anti-pSYN1 antibody conjugated to acceptor beads (1.25 nM and 5 µg/mL final, respectively) was prepared in assay buffer, and 10 µL was added to each well. The cell plates were briefly centrifuged and incubated at room temperature for 1 h. Donor beads (40 µg/mL final) were prepared in assay buffer, and 10 µL was added to each well. The plates were sealed with foil, briefly centrifuged, and incubated at room temperature for 1 h. Luminescence was detected using an EnVision multilabel plate reader (PerkinElmer) containing an AlphaScreen module. Compounds for the pilot screen and hit confirmation were acquired from several commercial vendors, including ChemBridge Corporation (San Diego, CA), Maybridge Chemicals (Thermo Fisher Scientific, Waltham, MA), InterBioScreen (Moscow, Russia), Enamine Chemicals (Ryan Scientific, Mt. Pleasant, SC), Biomol International (Enzo Life Sciences, Farmingdale, NY), Tocris Bioscience (Bristol, UK) and Sigma Aldrich Co., Ltd.
Viability Assay
Primary cortical neuronal cultures prepared from E17 Sprague Dawley rat embryos were seeded in black/clear 96-well poly-D-lysine–coated plates (BD Biosciences) at a density of 80,000 cells per well, and the total volume in each well was 80 µL. The cell plates were incubated at 37 °C in a humidified 5% CO2 incubator, and 50 µL of glutamate-free culture medium was added to each well 3 to 5 days later. Twenty days after seeding, the medium was removed and replaced with 100 µL of fresh glutamate-free culture medium containing compound, and the cell plate was incubated for 24 h at 37 °C. AlamarBlue reagent (Invitrogen) was added, and the cell plate was incubated for 1 h at 37 °C. Fluorescence was detected using an EnVision multilabel plate reader.
Data Analysis
Z factor was calculated using intraplate screening control values and the following equation: Z′ = 1 – (3*STDEV of 0% pSYN1 dephosphorylation + 3*STDEV of 100% of pSYN1 dephosphorylation)/(Average of 100% pSYN1 dephosphorylation – Average of 0% pSYN1 dephosphorylation), where 0% and 100% pSYN1 dephosphorylation indicate responses from vehicle (1% DMSO) and ionomycin-treated wells, respectively. Percent of pSYN1 dephosphorylation by compound was calculated using the following equation: % pSYN1 dephosphorylation by compound = 100 × (Average of 0% pSYN1 dephosphorylation – AlphaScreen signal of compound)/(Average of 0% pSYN1 dephosphorylation – Average of 100% pSYN1 dephosphorylation). Data analyses for concentration-response experiments were performed using GraphPad Prism 4 (GraphPad Software, La Jolla, CA), and EC50 values were derived from data fitted using a sigmoidal concentration response (variable slope) model.
Results and Discussion
Characterization of Primary Neuronal Cultures for HTS
To develop an HTS assay to detect pSYN1, we first established optimal primary neuronal culture conditions for the 384-well format. To confirm that the primary neurons undergo robust synapse formation when cultured in 384-well plates, immunostaining of the primary cortical neurons was performed to visualize the distribution of endogenous pSYN1. Cultures were triple-stained for pSYN1 (red), microtubule-associated protein 2 (MAP2, a neuronal dendritic marker) (green), and intact cell nuclei (blue, Hoechst stain). Immunostaining of cultures at 20 days in vitro (DIV) revealed a robust neural network marked by dendritic compartments containing MAP2 and mature synapses expressing pSYN1 ( Fig. 1 ). These results confirm that the cultures exhibit typical neuronal morphology, undergo extensive synaptogenesis, and express pSYN1 when grown under conditions suitable for HTS.
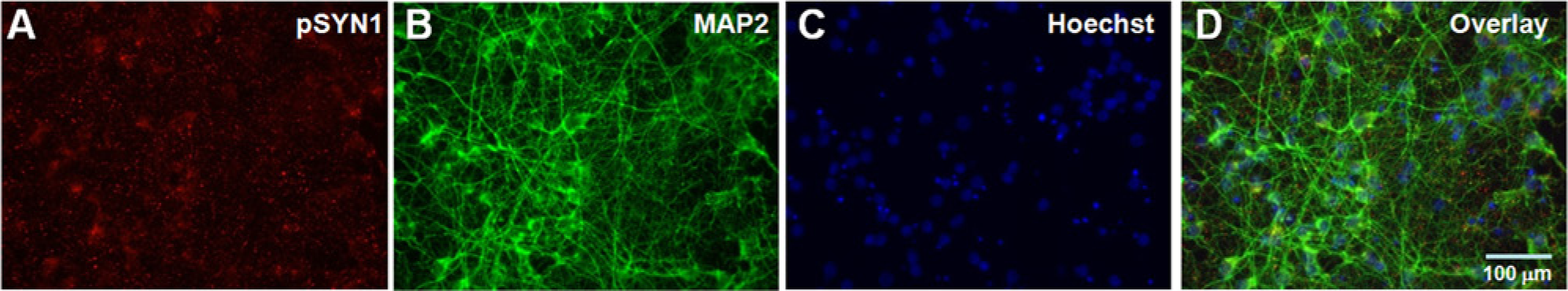
Triple-labeled immunofluorescence of primary cortical neurons. Primary cortical neurons were cultured in 384-well plates for 20 days. The cells were fixed and stained for (
Development of an AlphaScreen pSYN1 Assay
We selected the AlphaScreen system as a potential basis for a neuronal phosphoprotein detection HTS assay based on several key features. AlphaScreen (amplified luminescent proximity homogeneous assay) technology is a nonradioactive, proximity-based, sandwich immunoassay that can be initially optimized as a single-well cell-based assay that is amenable to further optimization for HTS.
19
The system uses donor and acceptor beads coupled to detection reagents that can be customized for a specific assay. Laser excitation of the donor beads at 680 nm converts oxygen (O2) to excited singlet oxygen (1O2), and a reaction cascade on proximal acceptor beads results in a light emission that can be measured at 615 nm.
19
For the pSYN1 assay, we coupled an anti-pSYN1 antibody to acceptor beads and employed streptavidin-coated donor beads to capture a biotinylated anti–total synapsin I antibody (
The feasibility of applying AlphaScreen technology was first evaluated using cell lysates. Primary cortical neurons cultured for 20 DIV were treated for 10 min with vehicle (1% DMSO) or 10 µM ionomycin, which is a calcium ionophore that activates calcineurin by increasing intracellular levels of calcium. Western blot analysis of these lysates showed complete dephosphorylation of pSYN1 after ionomycin treatment ( Fig. 2A ). Critically, a robust signal could also be detected using AlphaScreen ( Fig. 2B ), suggesting that the AlphaScreen platform has sufficient sensitivity for further optimization and miniaturization for development of an HTS assay to monitor pSYN1 in primary neurons.
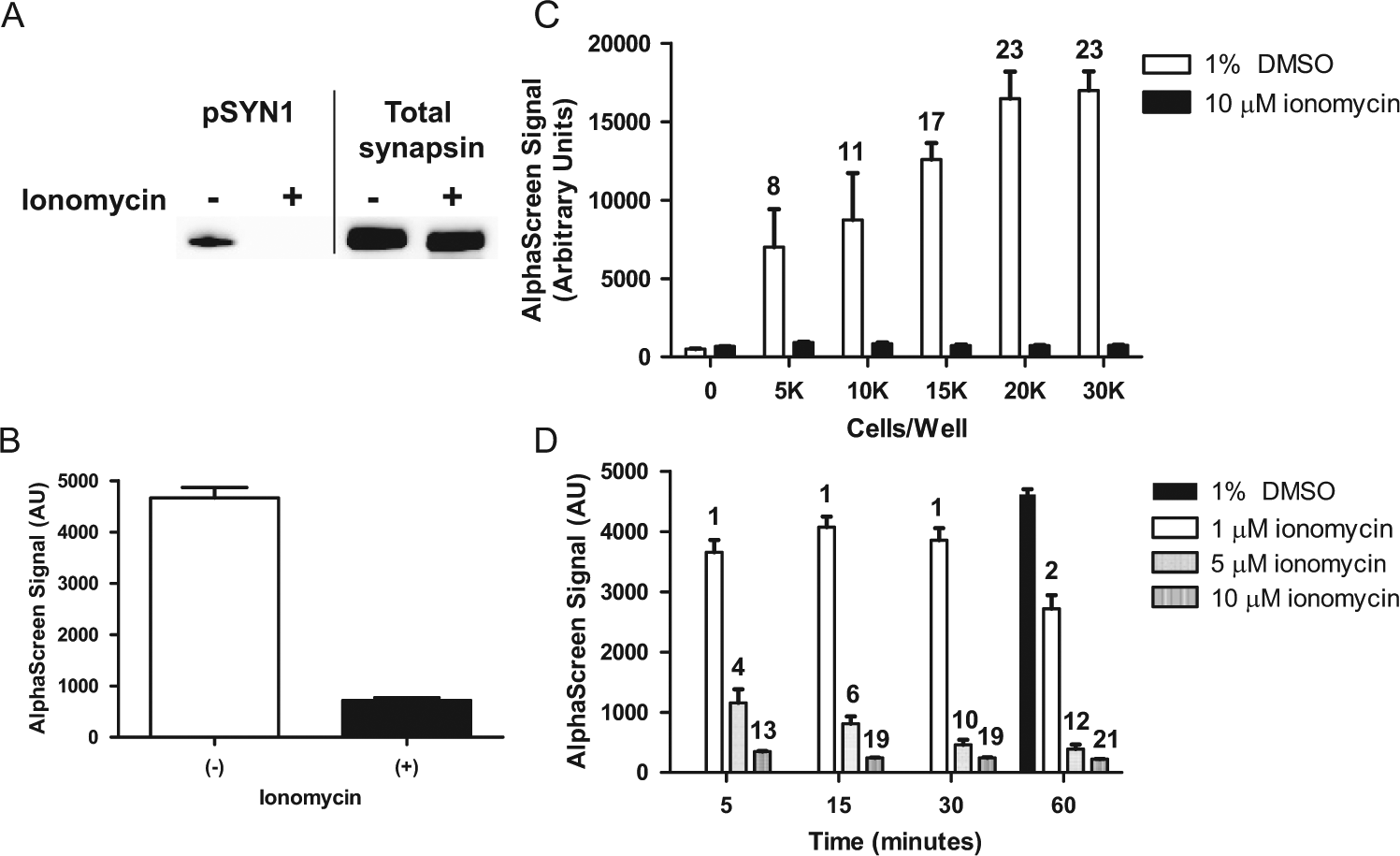
Detection of pSYN1 after ionomycin treatment. (
We next determined the optimal cell number per well to generate a robust assay window. Primary cortical neurons were seeded in 384-well plates at 5, 10, 15, 20, and 30 × 103 cells per well. After 20 DIV, the cells were treated with 1% DMSO or 10 µM ionomycin for 10 min. The magnitude of baseline pSYN1 signal in vehicle-treated wells increased with cell number and reached a plateau at 2 × 104 cells per well density ( Fig. 2C ). Treatment with ionomycin resulted in reduced pSYN1 levels at all culture densities. The assay window was defined by full activation of pSYN1 dephosphorylation by ionomycin, and a 23-fold difference was observed between vehicle and ionomycin-treated samples at 2 × 104 cells per well. These results suggest that this density is optimal for this assay.
Titrations of beads and biotinylated anti–total synapsin I antibody were carried out to determine optimal concentrations to yield the maximal signal-to-background ratios (
To determine the optimal concentration and incubation period for ionomycin as a positive control to be employed in HTS, a concentration-response and time course of ionomycin-induced pSYN1 dephosphorylation was performed in 384-well plates. Cells were treated with 1, 5, or 10 µM ionomycin for 5, 15, 30, or 60 min ( Fig. 2D ). Stimulation with 1 µM ionomycin induced partial pSYN1 dephosphorylation after 5, 15, and 30 min of treatment, and further dephosphorylation was observed after 60 min. Treatment with 5 µM ionomycin resulted in a significant decrease of pSYN1, which was also time dependent. A ~20-fold reduction in pSYN1 was observed with 10 µM ionomycin after 15 min, and no further dephosphorylation was detected with prolonged incubations, suggesting that maximal dephosphorylation was achieved in this period.
Ionomycin has been shown to induce neurotoxicity and protein degradation.
20
To exclude the possibility that the reduction of pSYN1 signal was due to protein degradation, we performed Western blot analysis to evaluate levels of pSYN1 and total synapsin I protein (
To determine optimal incubation periods and to evaluate the stability of the signal generated from the interactions between detection reagents and pSYN1, we carried out time course analyses. After lysing untreated primary cortical cultures at 20 DIV, biotinylated anti–total synapsin I antibody and anti-pSYN1 antibody conjugated to acceptor beads were added to the lysates. The plates were incubated at room temperature for 20, 45, 60, or 120 min. Donor beads were added, and the plates were incubated for an additional hour ( Fig. 3A ). A time-dependent linear increase in pSYN1 signal was observed, which reached a plateau at 60 min, and the signal was stable up to 2 h of initial incubation. In a separate experiment using untreated cells, the lysates were incubated with antibodies mixture for 1 h. Then donor beads were added, and the plate was incubated for 0.5, 1, 2, 3, 4, or 18 h ( Fig. 3B ). The pSYN1 signal reached maximal levels at 1 h and started to decrease with longer incubation periods. On the basis of these results, we decided to perform the assay using 1-h incubations.
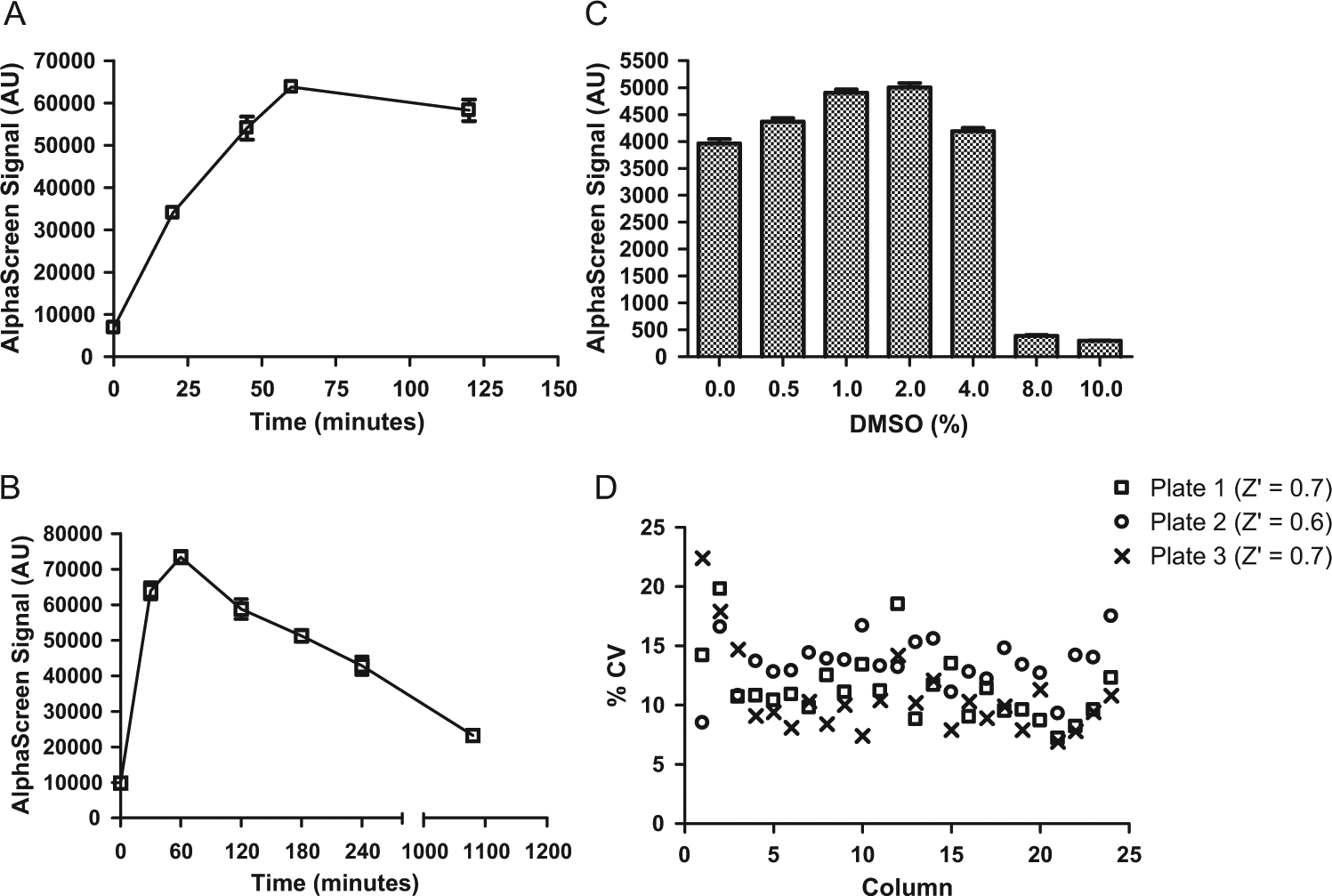
AlphaScreen assay optimizations. (
Since DMSO is typically used to solubilize compounds, it was necessary to assess the DMSO tolerance of the AlphaScreen assay. Cells were treated with 0%, 0.5%, 1%, 2%, 4%, 8%, and 10% DMSO for 1 h and the pSYN1 signal evaluated by AlphaScreen. The results of this analysis suggested that exposure of primary neurons and detection reagents to DMSO concentrations up to 4% does not affect the levels or detection of pSYN1 ( Fig. 3C ). We chose to perform the assay in 1% DMSO for compound screening.
Evaluation of Assay Variability
Well-to-well and plate-to-plate variabilities were examined to assess plate homogeneity and assay reproducibility, respectively. Primary cortical neurons were treated with 1% DMSO (columns 1–23) or 10 µM ionomycin (column 24) for 1 h, and the AlphaScreen pSYN1 assay was performed on three different days. The coefficient of variation (CV) for each column from the three plates was calculated ( Fig. 3D ). Low signal corner effects were observed, which were likely due to evaporation during the 20-day incubation period. When the corner wells were omitted from analyses, the CV values ranged from 7% to 15%, and the overall CV was <20%, an acceptable variance level for cell-based assays. 21 The Z factor, an indication of assay robustness, 22 was calculated using data obtained from columns 23 and 24 (corner wells were omitted). Z factors were determined to be 0.6 or greater, demonstrating that this assay is suitable for HTS.
Validation of the AlphaScreen pSYN1 Assay
Since phospho-S549 (site 6) of synapsin I is phosphorylated by the kinase CDK5, 10 a CDK5 inhibitor is expected to reduce levels of pSYN1 at this site. Roscovitine (Sigma), a nonspecific CDK inhibitor, 23 was used as a reference compound to validate the AlphaScreen pSYN1 assay. Treatment with roscovitine resulted in a concentration-dependent decrease of pSYN1 levels ( Fig. 4A ). The EC50 value obtained with roscovitine in this cellular assay is within the range of those described for other cellular effects of this compound. 23 This result confirms the capacity of the AlphaScreen pSYN1 assay to detect modulators of synapsin I phosphorylation at site 6.
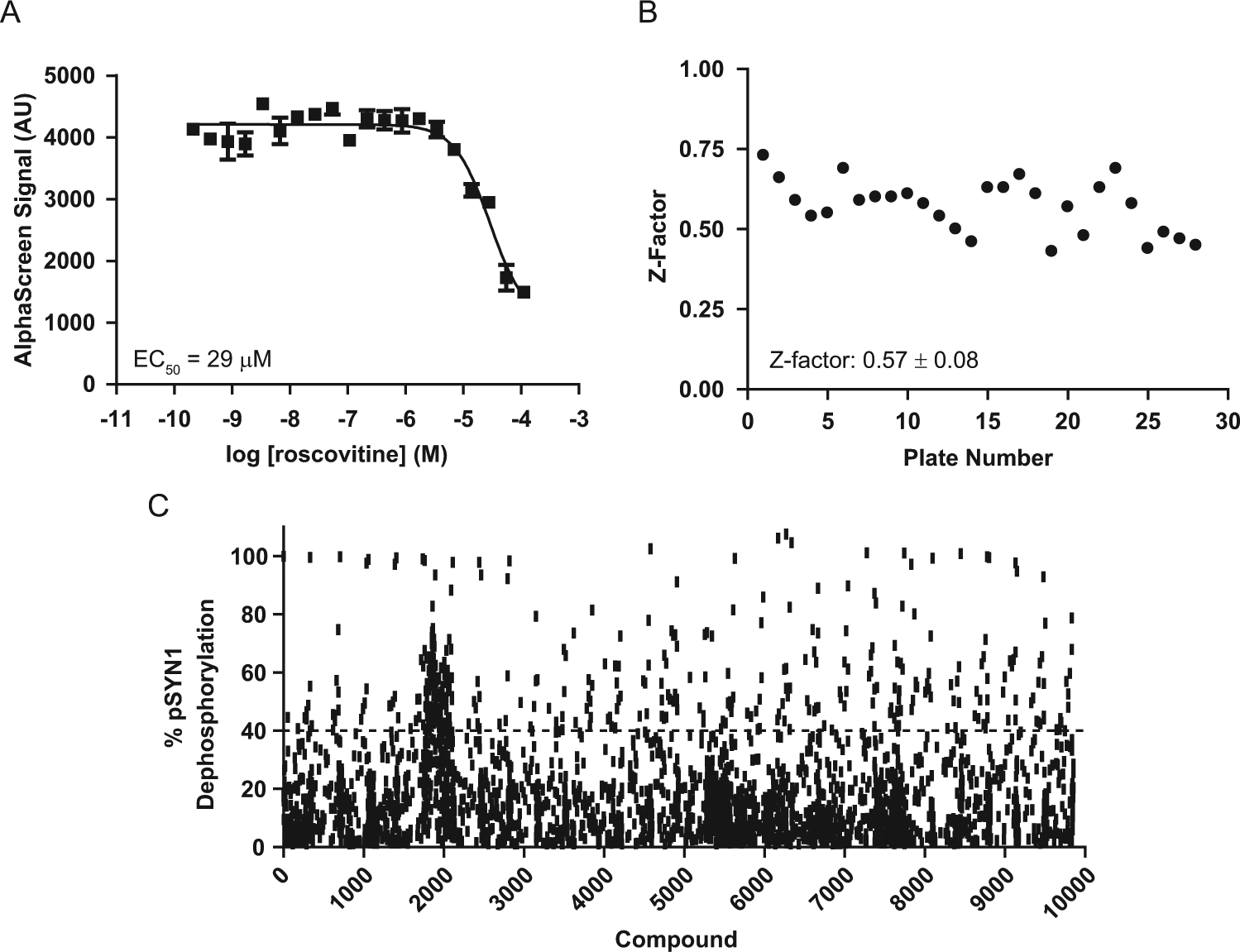
pSYN1 AlphaScreen assay validation and pilot screen performance. (
Pilot Screen Performance and Results
A pilot screening campaign was performed in the 384-well format to identify compounds that mediate pSYN1 dephosphorylation at site 6 in primary neurons. Twenty-eight 384-well plates (~10,000 compounds) were screened at 10 µM, and the final DMSO concentration was 1%. Intraplate screen controls (1% DMSO and ionomycin) were used to calculate Z factors. The pilot screen demonstrated good performance with a Z factor ranging from 0.46 to 0.73 with a mean ± SEM of 0.57 ± 0.08 ( Fig. 4B ), a robust value for an intricate 384-well screening format using primary neurons. Percent of pSYN1 dephosphorylation by compound was calculated by normalizing against DMSO and ionomycin controls (0% and 100% pSYN1 dephosphorylation, respectively) for each plate. pSYN1 dephosphorylation of 40% was chosen as a hit detection threshold, and 70 compounds were identified by this criterion, representing a 0.8% initial hit rate ( Fig. 4C ). These compounds were rescreened in duplicate, and 21 were confirmed as hits in the AlphaScreen pSYN1 assay. Analysis of these compounds using biotinylated acceptor beads and streptavidin-coated donor beads (TruHits; PerkinElmer) demonstrated that these confirmed hits were not false positives resulting from interference with the assay reagents (data not shown).
Concentration-response studies were next performed with nine confirmed hits in quadruplicate using reordered powders (
Fig. 5A
). The EC50 values and Emax percentages (relative to ionomycin) of pSYN1 dephosphorylation ranged from 23 nM to 6 µM and 41.5% to 105%, respectively, among these hit compounds (
Table 1
). There was no correlation between the potency and efficacy of hit compounds. For example, compounds 2 and 4 exhibited ~50% activation with EC50 values of 2.3 µM and 23 nM, respectively. To confirm that reduction of pSYN1 levels by these compounds was not due to protein degradation or loss of cells from the plate surface during the assay, Western blot analysis was performed to evaluate the levels of pSYN1 and total synapsin I protein. Primary cortical neurons were treated with varying compound concentrations, including EC50 values, under conditions used for the pilot screen. Concentration-dependent decreases in pSYN1 were observed with no effects on levels of total synapsin I with all compounds except compound 3 (
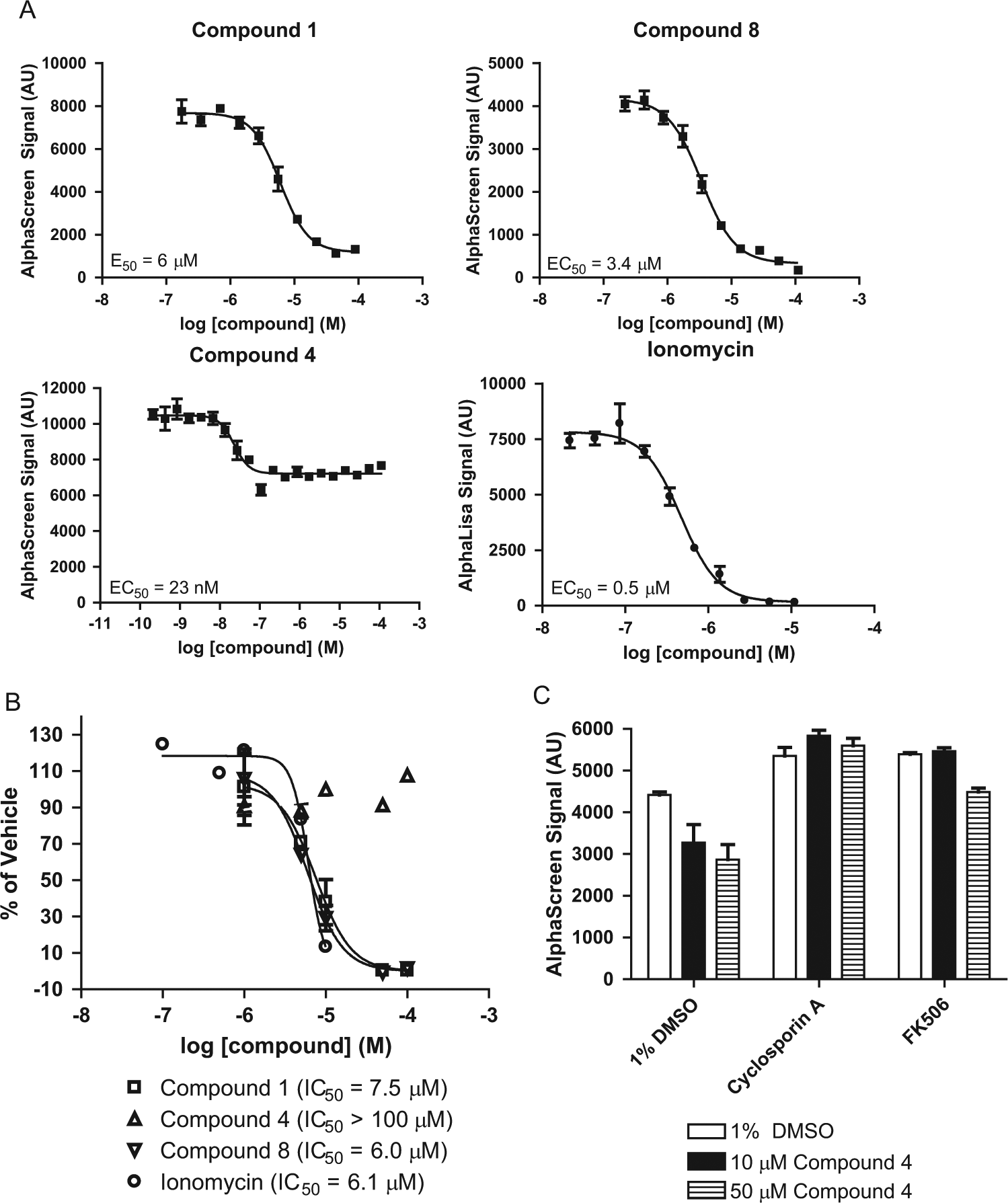
Characterization of compounds. (
Summary of Hits.
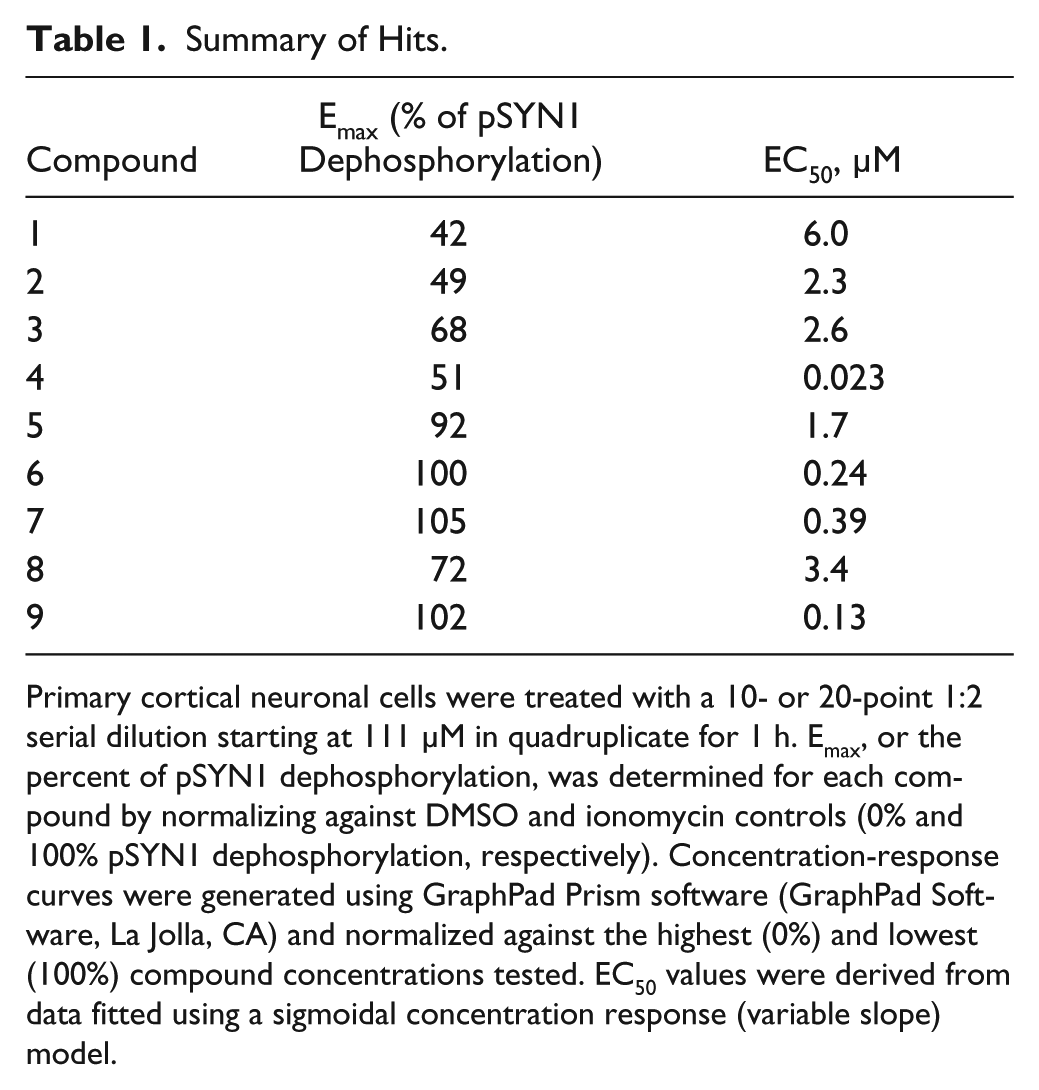
Primary cortical neuronal cells were treated with a 10- or 20-point 1:2 serial dilution starting at 111 µM in quadruplicate for 1 h. Emax, or the percent of pSYN1 dephosphorylation, was determined for each compound by normalizing against DMSO and ionomycin controls (0% and 100% pSYN1 dephosphorylation, respectively). Concentration-response curves were generated using GraphPad Prism software (GraphPad Software, La Jolla, CA) and normalized against the highest (0%) and lowest (100%) compound concentrations tested. EC50 values were derived from data fitted using a sigmoidal concentration response (variable slope) model.
Characterization of Hit Compounds
Initial characterization studies were carried out to determine mechanism of action of the nine compounds identified from the AlphaScreen pSYN1 assay. Compound 6 was identified as calcimycin, a divalent cation ionophore antibiotic. 24 Compound 7 was identified as veratridine, a sodium ion channel activator and neurotoxin. 25 Because of their known toxicities, further characterizations of these two compounds were not pursued. Compounds 8 and 9 are both members of the tyrphostin family of tyrosine kinase inhibitors, and their pharmacophores may be interesting for future investigations. These findings confirm the ability of the AlphaScreen pSYN1 assay to identify a diverse set of compounds.
Hit compounds were further evaluated to rule out undesirable cytotoxic effects. Primary neuronal cultures were incubated with compound for 24 h, and cell viability was tested using AlamarBlue (Invitrogen). Treatment with compounds 1 and 8 for 24 h resulted in a decrease in cell viability, and their cytotoxic effects were as potent as that of ionomycin. No reduction in cell viability was observed with compound 4 at all concentrations tested ( Fig. 5B ). Based on the large window between efficacy in pSYN1 dephosphorylation and cytotoxicity (>100 µM), compound 4 was selected as a promising hit for further analysis.
To determine whether compound 4 induced dephosphorylation of pSYN1 via a calcineurin-mediated pathway, primary cortical neuronal cells were pretreated with calcineurin inhibitors, cyclosporin A, or FK506 for 30 min, followed by treatment with compound 4. In the absence of compound 4 treatment, an increase in the level of pSYN1 was observed after treatment with cyclosporin A or FK506, suggesting the presence of basal calcineurin activity in the cultures ( Fig. 5C ). Critically, the calcineurin inhibitors blocked the ability of compound 4 to decrease pSYN1 levels, providing preliminary evidence that compound 4 mediates pSYN1 dephosphorylation via the calcineurin signaling pathway. Further investigation is necessary to confirm the role of compound 4 in this pathway.
In summary, we have demonstrated successful methods for culturing neurons to a stage of synaptic maturity in a high-throughput format and the development of a homogeneous, sensitive, and robust assay for detecting dephosphorylation of pSYN1 in primary neurons in 384-well plates. From a pilot screen, we identified several small molecules that modulate synapsin I phosphorylation (site 6) via multiple signaling pathways, including calcineurin. Further in vitro and in vivo characterizations and structure-activity relationship studies with compound 4 are currently in progress to understand its effects on neurotransmission and behavior. In addition, methods described here can be optimized for detecting modulators of other phosphorylated sites of synapsin I.
The ability to screen in primary neurons for compounds that modulate synapsin I phosphorylation has potential therapeutic implications. Dysregulation of pathways leading to synapsin I modification can result in abnormal phosphorylation states, which may lead to altered vesicle trafficking and neurotransmission in disease. Indeed, hyper- and hypophosphorylation states of synapsin I have been linked to Alzheimer and Huntington diseases.16,17 In addition, variations in a gene encoding a catalytic subunit of calcineurin, which is involved with dephosphorylation of synapsin I at site 6, have been reported to be genetically associated with schizophrenia,26,27 and forebrain-specific calcineurin knockout mice display a spectrum of behavioral abnormalities relevant to schizophrenia. 28 The implementation of the AlphaScreen pSYN1 assay may lead to the discovery of potential treatments for these disorders.
Moreover, multiple lines of evidence suggest that dysfunctions of synaptic transmission and, ultimately, neural circuit functions contribute to symptoms of neurodevelopmental and neurodegenerative diseases such as schizophrenia, autism, and Alzheimer disease. 29 Discovery of small molecules that restore normal neurotransmission is an attractive avenue for novel therapeutic interventions. Future development of primary neuronal assays that detect modifications of additional proteins involved in synaptic transmission may lead to the discovery and development of such critically needed therapies.
Footnotes
Acknowledgements
We thank Chantal Illy, Christian Fafard, and Greg Warner (PerkinElmer) for their technical support during the development and optimization of the AlphaScreen pSYN1 assay, as well as John Munro, Rasheedah Malik, and Daryl Johnson for their assistance with compound handling and plate preparations.
Declaration of Conflicting Interests
JRC, PL, and DJG are employees and stockholders of Galenea Corp. BC, BL, CJA, and JS are stockholders of Galenea Corp.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by a research and development collaboration with Otsuka Pharmaceutical Co., Ltd.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.