
Abstract
Alzheimer’s disease (AD) is a devastating neurodegenerative disease affecting millions of people. β-secretase-1 (BACE1), an enzyme involved in the processing of the amyloid precursor protein (APP) to form Aβ is a validated target for AD. Herein, the authors develop and validate a novel binding assay for BACE1 using the AlphaScreen platform that is amenable for high-throughput screening (HTS). Small-molecule BACE1 inhibitors of the hydroxyethylamine, hydantoin, and sulfamide classes were functionalized by biotin PEG linkers of varying lengths forming probes that were bound to streptavidin donor beads. BACE1 was coupled to nickel-chelate acceptor beads. Upon mixing, probes designed from all three classes registered high signal-to-background values in the AlphaScreen binding assay, where the interaction between probe and BACE1 was completely blocked by free parent compound. A probe from the hydantoin class was chosen for further optimization, where the final assay conditions of 50 nM BACE and 250 nM probe were used and Z′ values >0.75 were commonly observed. IC50 values determined by the AlphaScreen assay format exhibited ~10-fold greater sensitivity when compared with a fluorescence polarization–based activity assay. The assay was miniaturized to a 1536-well format for HTS, in which 525 000 compounds were screened.
Introduction
Alzheimer’s disease (AD) is a chronic and progressive neurodegenerative disease characterized by irreversible loss of memory, overall cognitive decline, and ultimately death. It is estimated that more than 20 million people worldwide have AD, where it is the leading cause of dementia in the elderly population. 1 Several defining hallmarks of AD include the presence of senile plaques consisting of insoluble Aβ, neurofibrillary tangles composed of hyperphosphorylated tau, and dystrophic neurites. 2 Biochemical, genetic, and animal model data suggest that the pathogenesis of AD is related to the accumulation of Aβ1-40 and Aβ1-42, more formally known as the amyloid hypothesis. 3 Aβ1-40 and Aβ1-42 are formed by the sequential proteolytic cleavage of the amyloid precursor protein (APP) by β-secretase-1 (BACE1) and γ-secretase.2–6
BACE1 is a type I integral membrane glycoprotein consisting of a large ectodomain of ~434 amino acids, a single transmembrane domain of ~22 amino acids, and a short cytoplasmic tail of 24 residues. BACE is an aspartyl protease in which the active site contains two conserved aspartic acid residues at Asp32 and Asp228 within the ectodomain. BACE has two isoforms (BACE1 and BACE2), where BACE2 has 68% similarity to BACE-1.5,6 Despite the high homology to BACE1, BACE2 has a different tissue distribution, different subcellular localization, and substrate specificity compared with BACE1.5–7 In a series of genetic knockout experiments in mice, it was shown that APP does not get processed to Aβ in the absence of BACE1, indicating that BACE1 is the isoform of relevance for AD.5,6,8
In the opinion of many investigators, BACE1 is a validated target for AD. Mutations in APP that cause familial AD are all located near the secretase cleavage sites. 6 For example, the Swedish mutation of APP is located immediately adjacent to the BACE1 cleavage site of APP.9,10 Synthetic peptides that contain the Swedish sequence are cleaved with 60-fold higher catalytic efficiency by BACE compared with APP peptide substrates based on the wild-type sequence. 11 In accordance with this result, cells expressing the Swedish mutant of APP accumulate higher levels of Aβ than cells expressing wild-type APP.6,9,10 Interestingly, individuals with Down’s syndrome, caused by an extra copy of chromosome 21, which contains the APP gene, invariably develop AD-like pathology usually by the fourth decade of life. 12 This clinical observation is consistent with the amyloid hypothesis and putative role of BACE1 in AD as higher substrate levels of APP that occurs as a result of the extra copy of the APP gene would lead to higher levels of Aβ accumulation. Recently, a mutation in APP was shown to protect against AD and age-related cognitive decline in data obtained from a cohort of 1795 Icelanders. 13
Historically, the most compelling data for validation of BACE1 as a target for AD comes from a number of genetic knockdown studies of BACE1 performed in AD transgenic mice. In Tg2576 and Tg6799 mice, genetic ablation of BACE1 rescued Aβ−dependent hippocampal memory deficits.14,15 In addition, Aβ-mediated cholinergic dysfunction was also rescued in Tg2576 BACE knockout mice. Although initial reports demonstrated that BACE1 knockout mice were healthy and normal, later reports showed that genetic knockout of BACE1 in mice did result in mechanism-based toxicity. 16 In a landmark study by McConlogue et al., 17 this group demonstrated that partial knockdown of BACE1 had dramatic effects on Alzheimer plaque and synaptic pathology in PDAPP mice. Furthermore, the heterozygous mice were determined to be healthy and normal in numerous cohorts. Taken together, the partial reductions of BACE1 enzyme activity that lead to a marked reduction of Aβ-mediated pathology in AD transgenic mice suggest that small-molecule inhibition of BACE1 could be disease modifying in the absence of a mechanism-based toxicity in patients.
There has been significant interest in BACE from the pharmaceutical industry, which has led to the development of a number of assays amenable to HTS. One specific example would be a fluorescence resonance energy transfer assay in which donor and acceptor fluorophores can be covalently attached to a BACE1 substrate peptide. Upon BACE1-mediated cleavage of the peptide, donor quenching is relieved with a concomitant emergence of acceptor fluorescence. 18 This type of assay can suffer from fluorescence interference from compounds commonly found in HTS libraries 19 and often times may not have a high enough signal-to-background (S/B) ratio for high-throughput screening (HTS). Fluorescence polarization (FP) assays have been used for other aspartic proteases 20 but also may suffer from compound-based fluorescent interference 21 as well as sensitivity issues. BACE1 inhibitors initially identified by conventional HTS using the BACE1 ectodomain and assay conditions of pH 4.5 have not been successful in generating compounds suitable for clinical testing. 22 Subsequently, structure-guided drug design approaches initiated by a fragment-screening effort have yielded more tractable compounds and are in clinical phase testing.23–26 This new wave of small-molecule BACE1 inhibitors may bring hope to AD patients in the future, yet it remains to be determined if they will have potential toxicity or pharmacokinetic (PK) liabilities in clinical phase testing.
More innovative assay approaches for BACE1 offering robust sensitivity have been described. One such example is a β-galactosidase (β-Gal) enzyme fragment complementation assay. 27 This assay uses a cyclic β-Gal enzyme donor peptide containing a protease-selective cleavage sequence. 27 After cleavage by BACE, the peptide is linearized and can now complement with the β-Gal enzyme acceptor forming an active β-Gal enzyme. However, it remains to be determined whether these other assay approaches will lead to compounds that are successful in clinical phase testing. Thus, new and better scaffolds for BACE1 inhibitors should be useful in the future. Herein, we describe a novel binding assay that is amenable to HTS to identify small-molecule inhibitors of BACE1. This assay uses the AlphaScreen technology and shows enhanced sensitivity compared with traditional assay formats.
Materials and Methods
Reagents
The recombinant BACE1 protein containing a C-terminal 6xHis tag was purchased from Proteos (Kalamazoo, MI) and stored at −80 °C in small aliquots until use. All small-molecule BACE inhibitors were synthesized in house, and NHS-PEG(n)-biotin was purchased from Thermo Scientific (Rockford, IL). The NHS ester (within NHS-PEG(n)-biotin) reacted with primary amine groups engineered in the BACE inhibitors to generate biotinylated probes, which were purified by high-performance liquid chromatography to >98% purity. The AlphaScreen histidine detection kit containing nickel-chelate acceptor and streptavidin-conjugate donor beads was acquired from PerkinElmer (Waltham, MA). The peptide substrate (Biotin-GLTNIKTEEISEISYEVEFRC [Oregon Green] KK-OH) used in the FP-BACE assay was custom made by Anaspec (Fremont, CA). Bovine serum albumin (BSA), Tween-20, streptavidin, and all other reagents were obtained from Sigma-Aldrich (St. Louis, MO). Polypropylene 96-well and polystyrene 384- and 1536-well microplates were purchased from Corning (Corning, NY).
AlphaScreen Binding Assay Development and Optimization
For initial assay characterization, BACE-6xHis (50 nM) and biotinylated probes (titrated from 10 nM to 10 µM) were mixed together in the assay buffer, phosphate-buffered saline (PBS) containing 150 mM NaCl, pH 7.4, 0.01% Tween-20, and 0.05% BSA in white 384-well low-volume microplates. Reactions were incubated for 1 h at 25 °C, nickel-chelate acceptor and streptavidin donor beads were then added simultaneously to a final concentration of 20 µg/mL each and incubated for 2 h. The plates were subsequently read on the EnVision multilabel reader (PerkinElmer) using the default settings (a 640 nm dichroic mirror for the excitation light and a 570 ± 50 nm cutoff filter for emission). To confirm the specificity of positive assay signals generated by the interactions between BACE-6xHis and biotinylated probes, we performed competition experiments with a high concentration of parent inhibitors. BACE-6xHis (50 nM) was preincubated with the parent inhibitors (100 µM) for 15 min, followed by the addition of respective biotinylated probes (1 µM). After incubation at 25 °C for 1 h, acceptor and donor beads were then added to a final concentration of 20 µg/mL and read on the EnVision using the AlphaScreen protocol after a 2 h incubation.
The assay buffer was further optimized to increase the S/B ratio. The concentrations of BACE-6xHis and the chosen biotinylated probe were maintained at 50 nM and 1 µM, respectively, and a series of experiments were performed on three different reagents that could affect assay signal: pH (6.5–7.4), NaCl (25–150 mM), and Tween-20 (0.001%–0.05%). In addition, DMSO was separately titrated in the optimized buffer (PBS with 150 mM NaCl, pH 7.0, 0.01% Tween-20, and 0.05% BSA) to examine its effect on assay signal. To confirm that the optimized assay buffer delivered a robust signal and low background, we performed further titration of BACE-6xHis and the chosen biotinylated probe. Selected for final assay validation and inhibitor screening were 50 nM BACE-6xHis and a 250 nM biotinylated probe.
Assay Automation and Miniaturization
The 384-well assay was performed using Matrix electronic multichannel pipettes and/or a Multidrop Combi reagent dispenser (Thermo Scientific, Hudson, NH). Four microliters of test compounds in 20% DMSO, equivalent DMSO controls (maximum signal), or 2 mM EDTA controls (maximum signal) were first added to the plates. Four microliters of BACE-6xHis was then added and followed by 4 µL of the biotinylated probe. The plates were incubated for 1 h at 25 °C, then 4 µL of a mixture of donor and acceptor beads was added to a final concentration of 20 µg/mL each and a final volume of 16 µL. The plates were incubated for 2 h at 25 °C before reading. The interday and interplate variation of the automated assay were examined over the course of 3 d by running two identical 384-well plates per day, in which the minimum and maximum signal control wells were positioned in alternating columns. We further miniaturized the assay to a 1536-well format by proportionally reducing the total assay volume in half to 8 µL.
Quantitative HTS
The library used in these studies represents an internal collection of approximately 525 000 compounds, consisting of both commercially available and Elan proprietary entities.
For the screen, the screening collection was transferred to 1536-well plates (Corning, cat No. 3725). Columns 5 to 44 contained compounds to be tested, and columns 1 to 4 and 45 to 48 contained negative (DMSO) and positive (5 µM compound) controls. Compounds were screened at a final concentration of 500 µM in 5% DMSO. Primary screening consisted of single-point screening, followed by hit confirmation in triplicate, as well as artifact screening to remove AlphaScreen-specific false-positives. Relative fluorescent units were normalized to the control compound to calculate a percentage of control (%POC) for each compound, using standard analysis calculations. Compounds were considered hits when the %POC was greater than the mean + 3 median absolute deviation (MAD).
The primary HTS hits were counterscreened in a 1536-well assay to identify false-positive hits that interfere with the AlphaScreen detection method. The compounds were plated as described above, and biotin-6xHis peptide (PerkinElmer) was then added to a concentration of 5 nM and incubated for 30 min at 25 °C prior to the addition of detection beads at 20 µg/mL. The reactions were incubated for 2 h at 25 °C before being read on the EnVision. The maximum control wells contained an equivalent volume of DMSO, whereas the minimum control wells had 0.5 mM EDTA.
FP-Based BACE Assay
An FP-based BACE was employed to determine the functional potency of BACE inhibitors in black 384-well microplates. Briefly, BACE-6xHis (0.1 nM final concentration) was preincubated with serially diluted inhibitors or DMSO control (5% final) in 20 µL assay buffer (100 mM sodium acetate, pH 4.5, and 0.001% Tween-20) for 30 min at 25 °C. The reaction was initiated with the addition of 10 µL of a biotinylated peptide substrate (450 nM in assay buffer) and incubated at 37 °C for 3 h. The reaction was subsequently quenched with the addition of 30 µL streptavidin (1.5 µM in a pH-neutral assay buffer). After incubation for 15 min at 25 °C, the plates were read on the EnVision reader.
Data Analysis
All experimental data were analyzed and plotted using Prism software (GraphPad Software, San Diego, CA). Curve fitting for IC50 values was done using four-parameter fits with variable slopes. The quality and robustness of the assay were determined by analysis of the Z′ factor. 28
Results
Characterization of BACE-Probe Interaction
Historically, small-molecule BACE inhibitors have been discovered primarily through HTS campaigns using functional biochemical assays and more recently through fragment screening and structure-guided drug design approaches.23–26 However, PK and safety issues as well as appropriate patient stratification have presented challenges toward advancing BACE inhibitors to late-stage clinical testing. Here, we investigated the use of an AlphaScreen binding assay to discover novel low-affinity BACE inhibitors. AlphaScreen is designed to measure the proximity of donor and acceptor beads conjugated to biomolecules of interest. When excited at 680 nm, the donor beads convert ambient oxygen to singlet oxygen, which diffuses out and comes into contact with the acceptor beads within 200 nm distance. The singlet oxygen then initiates a cascade of chemiluminescent reactions in the acceptor beads that emits light in the 520 to 620 nm range. Each donor bead can produce ~60 000 singlet-oxygen molecules per second, resulting in tremendous amplification of the binding signals. Because of the avidity of the AlphaScreen beads, binding partners can appear to have a higher affinity for each other, which is not uncommon with bead-based assay technologies. This feature makes direct binding kinetics measurement unsuitable; however, it renders AlphaScreen an ideal assay platform for capturing lower-affinity (i.e., in the micromolar range) molecular interactions.
Taking advantage of this unique feature of the AlphaScreen assays, we investigated the binding of the BACE1 protein to a panel of small-molecule probes derived from known BACE1 inhibitors ( Table 1 ). Probes were synthesized as described in Supplementary Methods, in which small- molecule BACE inhibitors of the hydroxyethylamine (HEA), hydantoin (S1), and sulfamide (S2–S3) classes were functionalized with a biotin PEG linker of varying lengths ( Fig. 1A ). BACE was obtained in >95% purity by nickel affinity and size exclusion chromatography. A C-terminal 6x His tag was engineered and retained for binding to the nickel-chelate acceptor beads. The streptavidin-conjugated donor beads interact with the biotin moiety of the probes. The formation of a complete detection complex, which contains BACE1, probe, and the donor and acceptor beads, should generate an AlphaScreen assay signal ( Fig. 1B ).
Profile summary of the 11 BACE probes generated and tested.
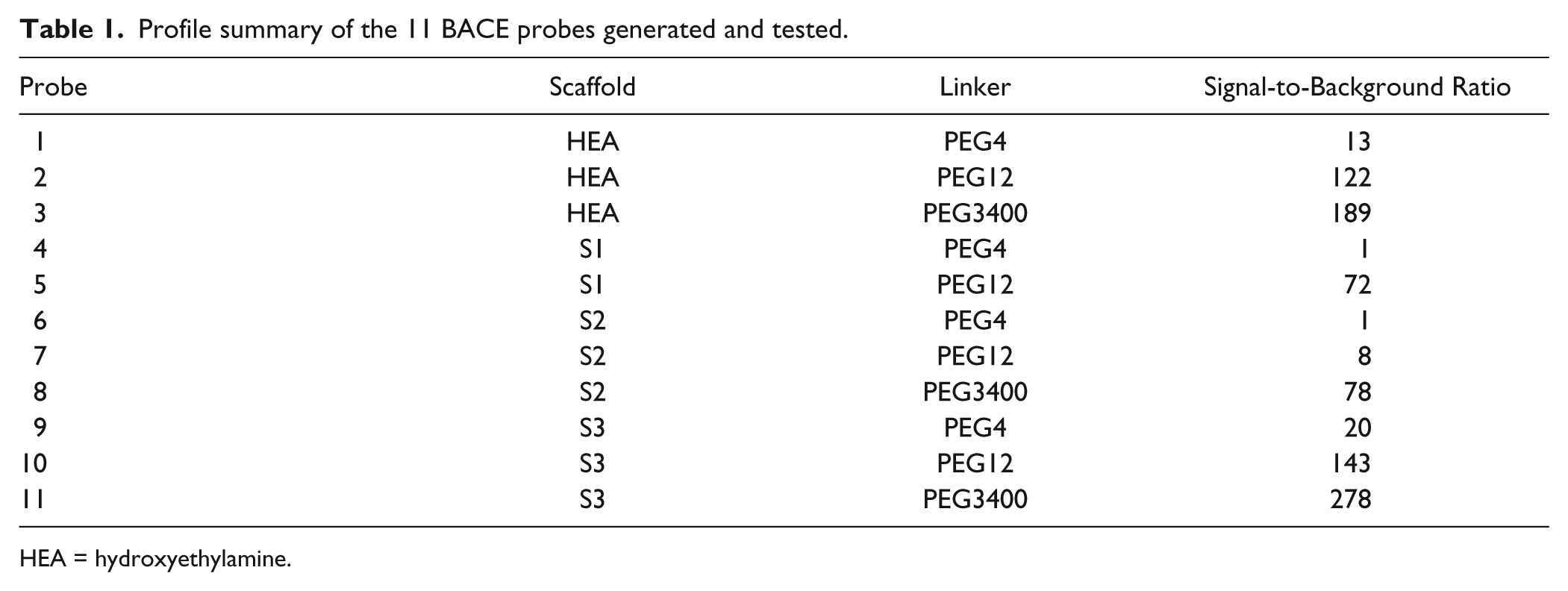
HEA = hydroxyethylamine.
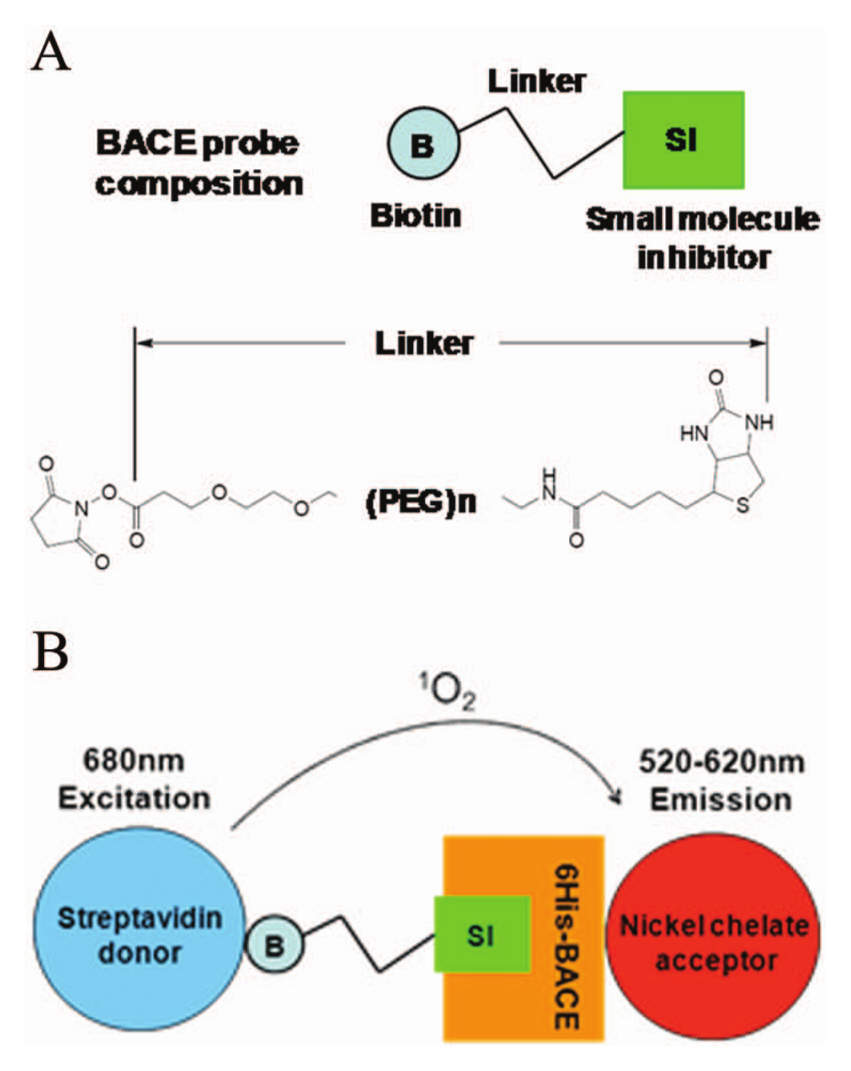
AlphaScreen binding assay platform for the discovery of novel BACE inhibitors. (
In our initial proof-of-concept study, we examined the binding profile of three probes (i.e., with viable PEG linkers) derived from a single high-affinity (100 nM) BACE1 inhibitor that belongs to the HEA class (
Fig. 2A
). We performed a titration for all three probes with concentrations ranging from 10 nM to 10 µM, while maintaining the BACE1 concentration constant at 50 nM. As shown in
Figure 2A
, positive binding events were detected for all three probes, with maximal signals observed at 1 µM for probe 1 and 2 and 0.1 µM for probe 3. The latter also generated the highest signal with an average S/B ratio of 190. In addition, a prominent hook effect was observed (
Fig. 2A
). This phenomenon is fairly common in homogeneous assays, in which an excess of individual components can result in unproductive binding that significantly decreases assay signal and/or increases background. To rule out the possibility that the observed binding events might be due to nonspecific interaction between the probes and BACE protein, we conducted a competition experiment in the presence of a saturating concentration of parent inhibitor or DMSO control. The robust signals generated from both HEA-derived probes were reduced to background level by the parent inhibitor, thus confirming the specificity of the BACE-probe interaction (
Fig. 2B
). Next, we expanded the same experimental paradigm to additional probes derived from two different classes of low-affinity (2–20 µM potency) BACE inhibitors (hydantoin S1 and sulfamides S2–S3;
Table 1
). In a titration study similar to the one described above, we observed positive signals generated by one or more probes belonging to each of these three chemical classes (
Fig. 2C
). It is important to point out that robust interaction was observed at probe concentrations significantly lower than the reported affinity values. This is another demonstration of the ability of AlphaScreen, due to a bead-mediated avidity effect, to capture low-affinity molecular interactions difficult to detect in other assay platforms. The specificity of these interactions was subsequently confirmed in a competition study with their respective parent inhibitors (
Fig. 2B
). In addition, to exclude the possibility of nonspecific probe binding to BACE1 accounting for the signal, full displacement curves of free parent to probes 1, 2, 3, 5, 7, and 8 are shown in
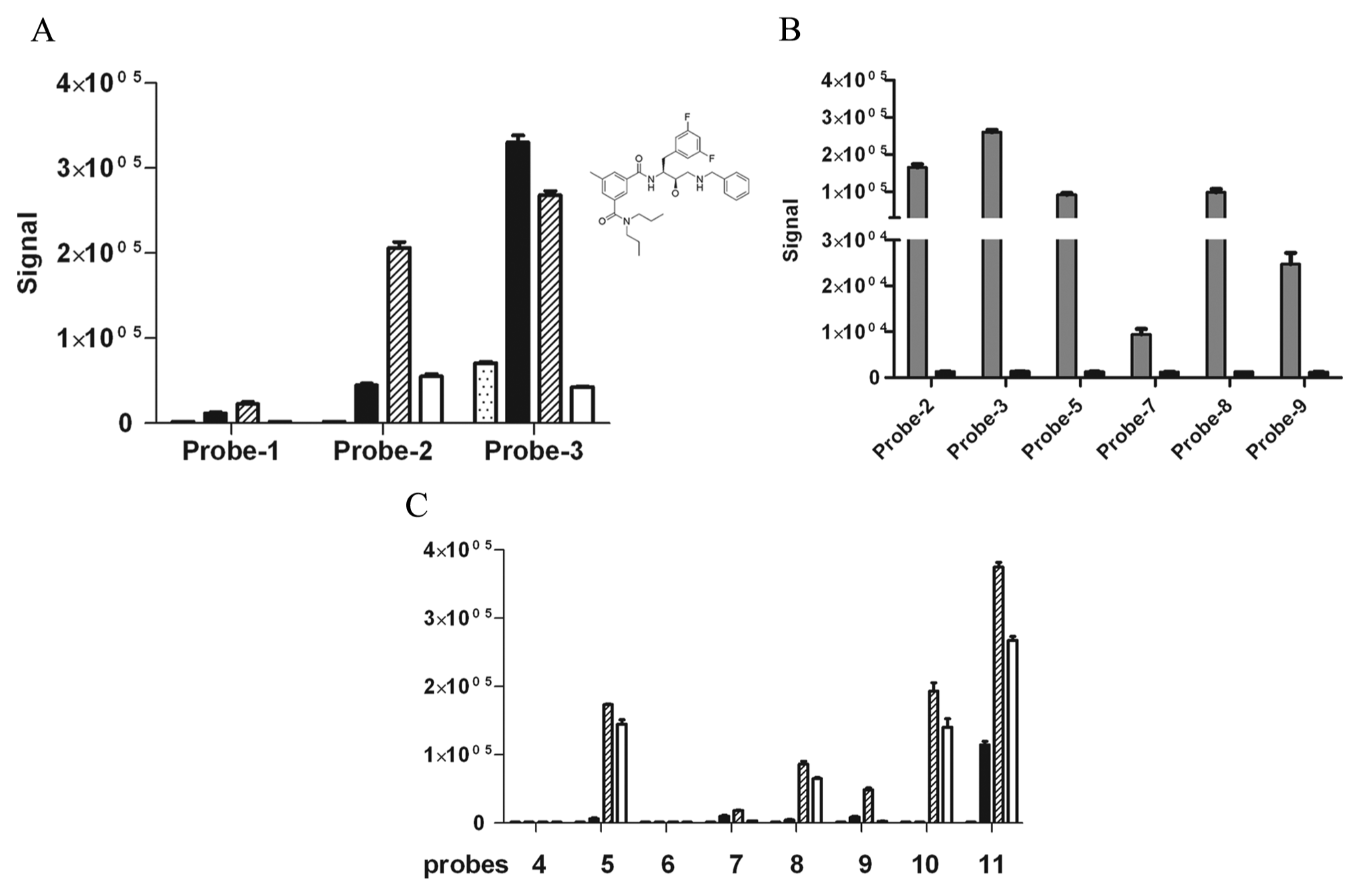
Evaluating probes from four different structure classes in the AlphaScreen binding assay. (
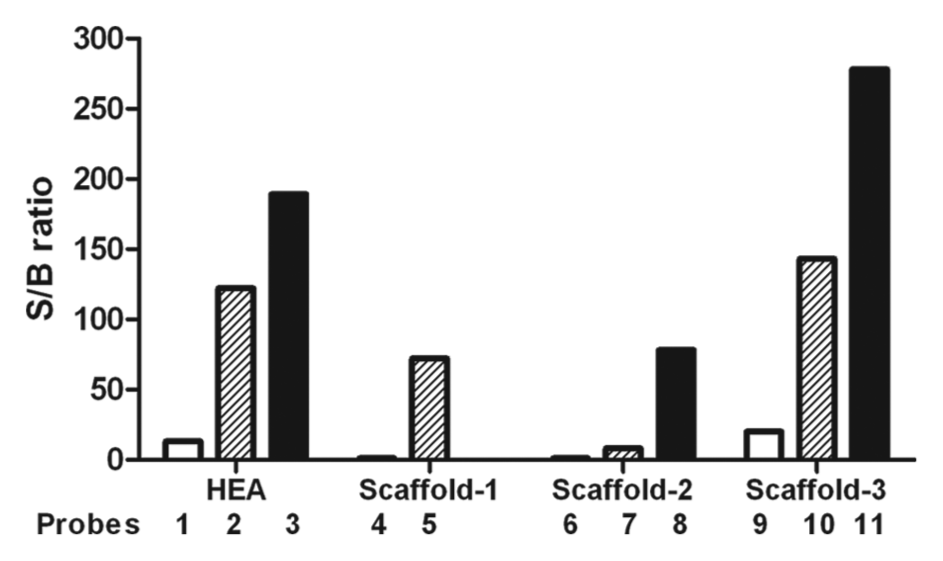
The effect of probe linker size on assay signal. Data generated from the probe-profiling study were further analyzed to illustrate the impact of linker size on assay performance. The data were collected at 1 µM probe concentration, with the exception of probe 3 (0.1 µM). Data are expressed as signal-to-background ratio, and the background is defined by negative controls treated with 0.5 mM of EDTA. Within each of the four structure classes tested, a longer linker size resulted in a higher assay signal in which PEG-3400 (black) > PEG-12 (hatched) > PEG-4 (white). Because of technical difficulties, a PEG3400-linked S1 probe could not be generated and purified for testing.
Assay Buffer Optimization for HTS
Among the three classes of low-affinity probes, we were most interested in exploring the pocket occupied by the S1 probes and discovering additional novel chemical entities that target the same binding site. To achieve that, a robust binding assay using probe 5 (S1 class with a PEG-12 linker) needs to be further optimized and validated before an HTS campaign can take place. First, we examined the impact of various buffer components on assay signals, including pH, Tween-20, NaCl, and BSA. As shown in
Optimizing BACE and Probe Concentrations
The optimized assay buffer was PBS pH 7.0, containing 150 mM NaCl, 0.01% Tween-20, and 0.05% BSA. To achieve the highest S/B ratio, we performed additional titrations of BACE1 and probe 5 in the presence of 5% DMSO. We first held the probe constant at 1 µM and tested a wide range of BACE1 concentrations. A rapid signal decline was detected when the BACE1 concentration was reduced from 100 nM to 0.05 nM, resulting in a complete loss of assay window with less than 10 nM BACE (
Fig. 4A
). We therefore chose a moderate BACE concentration of 50 nM for the subsequent probe titration. As shown in
Figure 4B
, a dose-dependent decrease of signal was also observed, with probe concentration ranging from 1 µM to 31 nM. Based on these titration data, we chose the combination of 50 nM BACE and 250 nM probe 5 as our final assay condition, balancing the need to conserve reagents while maintaining a robust assay window (~20-fold). To further evaluate the robustness and reliability of the assay for HTS, we studied interplate and interday variations with replicate plates containing both the minimum and maximum signal controls. The Z′ values were 0.81, 0.78, and 0.77 for each respective run, with interday variability in the maximum controls less than 10% (
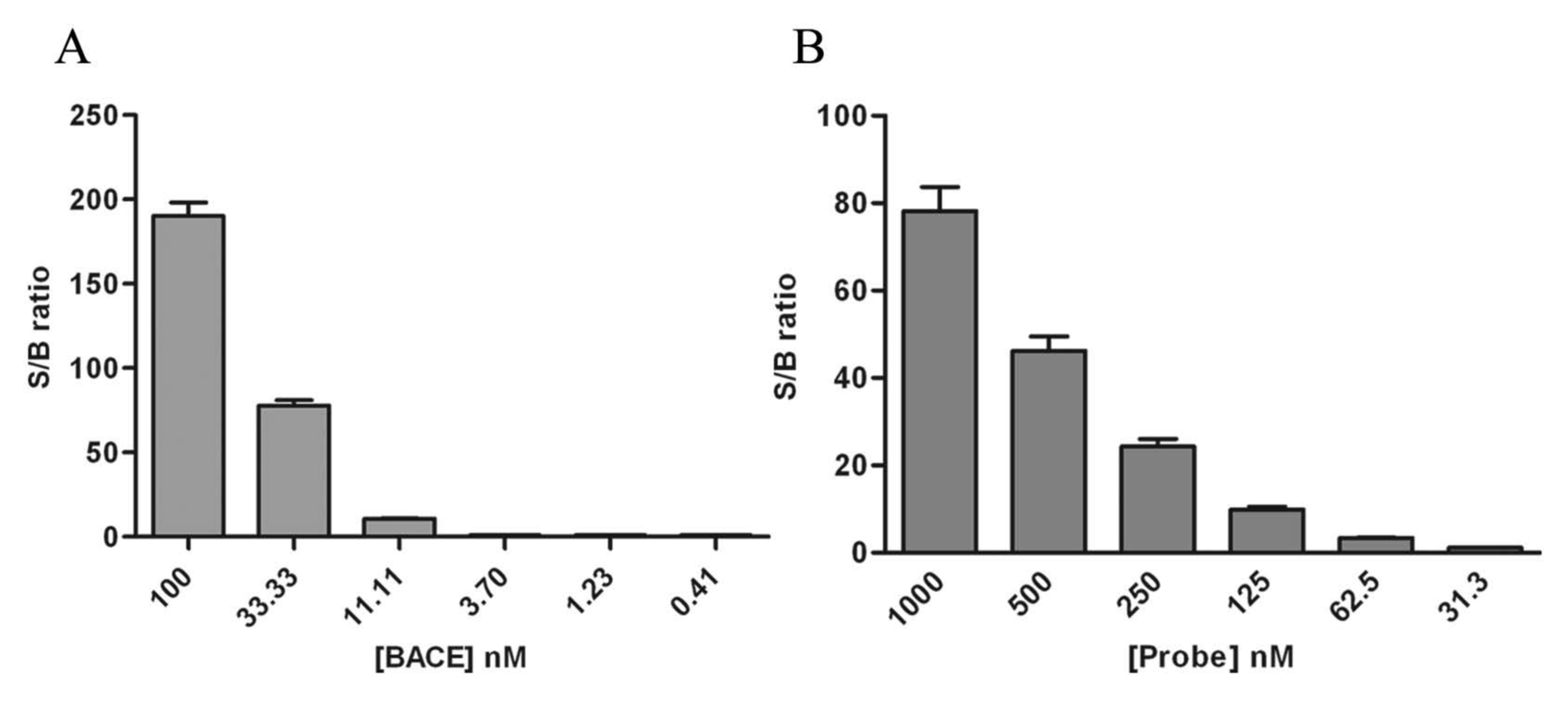
Titration of BACE and probe 5 in optimized assay buffer. (
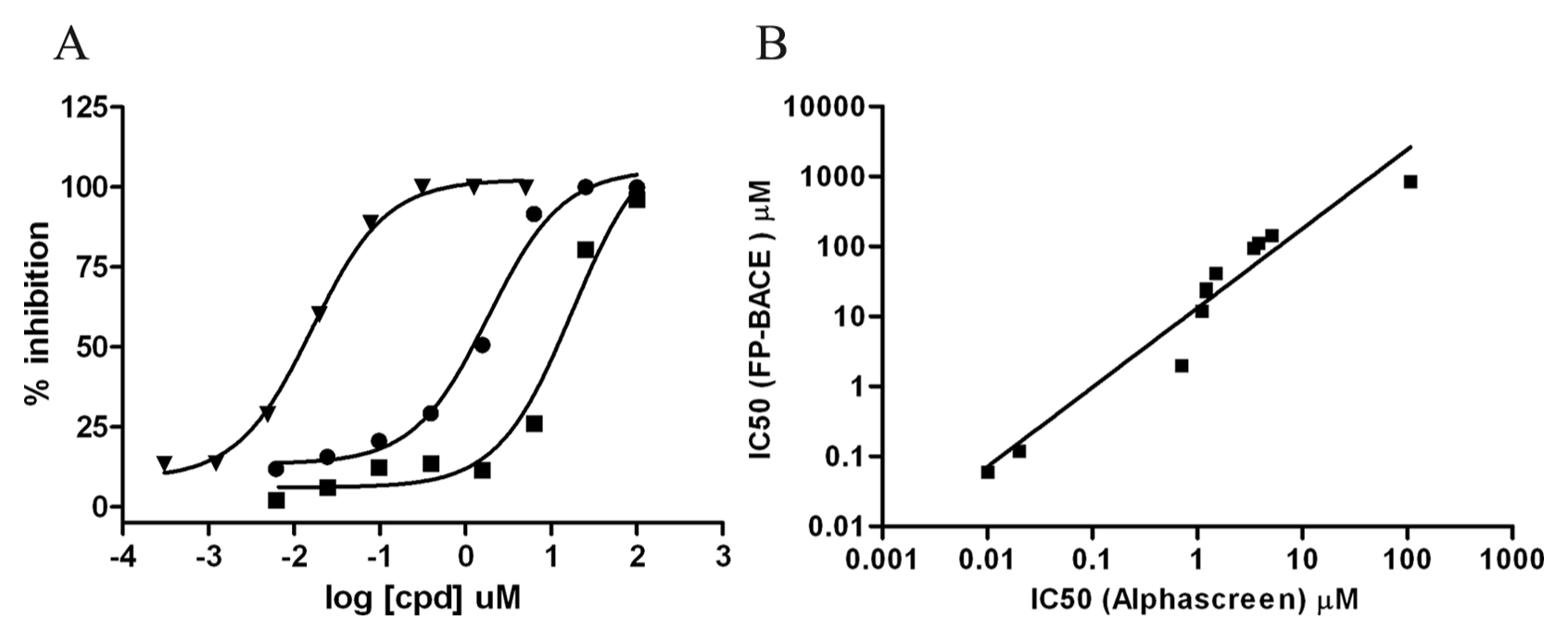
Characterization and validation of the AlphaScreen binding assay with BACE1 inhibitors. (
Assay Validation
Finally, to validate this novel binding assay pharmacologically, we tested a panel of 11 internal BACE inhibitors covering five different structure classes and compared their IC50 values to those obtained from an FP-based activity assay. As shown in
HTS of Internal Library and Hit Follow-up
We screened more than 525 000 compounds using a validated AlphaScreen assay miniaturized to a 1536-well format to identify BACE1 inhibitors. Because of the high screening concentration (500 µM), a high hit rate was predicted. Initial screening was followed in parallel by primary hit confirmation in triplicate and artifact counterscreening using percentage inhibition >24% (mean + 3 MAD) for hit calling, which identified 26 144 hits, yielding a final hit rate of 4.97% (
Biacore positive hits were then shuttled through high-throughput crystallography. Compound 12 (
Discussion
The binding assay and subsequent screen described in this article differ in many respects from that of previous assays used to screen and identify BACE1 inhibitors. In this assay, a BACE construct consisting of the BACE1 extracellular domain fused with a C-terminal 6x-histidine tag was coupled to nickel-chelate–coated acceptor beads. BACE1 inhibitors linked to biotin by PEG linkers of varying lengths (probes) were bound to streptavidin-coated donor beads. The interaction of the probe with the active site of BACE1 will bring the donor and acceptor beads into close proximity and thus yield a strong signal. Free compounds that will bind in the active site of BACE1 and displace the probe will abrogate the signal, forming the basis of our assay. Probes were based on three different classes, an HEA, a hydantoin, and two variants of sulfamides. For our actual HTS, probe 5, which was based on the hydantoin scaffold, was chosen as it does not displace water from the catalytic aspartic acids of BACE. This was important as energetically it is unfavorable to displace water from the catalytic aspartic acids, and this type of binding mode will usually increase the number of hydrogen bond donors of the small molecules, which can be unfavorable from a PK standpoint as well. HEA-based compounds will displace the water from the catalytic aspartic acid compounds, which could contribute in part to the poor PK characteristics that have been observed with some compounds in this class. However, it should be emphasized that this assay itself will identify compounds of different binding modes regardless of whether they displace water from the active site or not.
The optimal pH for measuring BACE1 activity using the extracellular domain construct is pH 4.5, whereas at a pH greater than 5.5, only minimal activity was observed (
A major advantage of the AlphaScreen assay described in this article compared with other assay formats is the enhanced sensitivity that is observed with this format, as shown in
The AlphaScreen format can tolerate higher DMSO concentrations, thus enabling the screening concentration (500 µM) used in this HTS campaign. We observed a relatively high hit rate of 4.97% (
The binding assay described for BACE1 can be extrapolated to other drug targets as well and could be considered a platform assay. A small-molecule binder can be functionalized with a biotin-PEG of varying lengths and coupled to streptavidin-coated beads. The target protein of interest can then be coupled to donor beads, and conditions of buffers and target concentrations can be optimized. Consistent with our results and those of others, this approach could enhance assay sensitivity by an order of magnitude, which could make screening possible for many targets that have nonoptimal Z′ values using more traditional assay approaches. Also, as this assay is a binding assay, it is amenable for targets in which functional activity cannot be measured or intrinsic enzyme activity is low in vitro.
Footnotes
Acknowledgements
We would like to acknowledge Pat Escaron, Balazs Szoke, and Paul Beroza for thoughtful discussions and Lany Ruslim for her technical expertise. We would also like to thank Cary Cochrell for his help in preparing the manuscript. We dedicate this article to Peter Seubert, thanks for believing.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Funding was provided by Elan Pharmaceuticals, and the research was performed as part of the ongoing work at Elan.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.