
Abstract
Tumor marker endothelial 8 (TEM8) is a receptor for the protective antigen (PA) component of anthrax toxin. TEM8 is upregulated on endothelial cells lining the blood vessels within tumors, compared with normal blood vessels. A number of studies have demonstrated a pivotal role for TEM8 in developmental and tumor angiogenesis. We have also shown that targeting the anthrax receptors with a mutated form of PA inhibits angiogenesis and tumor formation in vivo. Here we describe the development and testing of a high-throughput fluorescence resonance energy transfer assay to identify molecules that strongly inhibit the interaction of PA and TEM8. The assay we describe is sensitive and robust, with a Z’ value of 0.8. A preliminary screen of 2310 known bioactive library compounds identified ebselen and thimerosal as inhibitors of the TEM8-PA interaction. These molecules each contain a cysteine-reactive transition metal, and complementary studies indicate that their inhibition of interaction is due to modification of a cysteine residue in the TEM8 extracellular domain. This is the first demonstration of a high-throughput screening assay that identifies inhibitors of TEM8, with potential application for antianthrax and antiangiogenic diseases.
Introduction
Tumor endothelial marker 8 (TEM8) is a von Willebrand factor domain-containing receptor protein with high structural homology to integrin receptors. 1 The receptor has been most thoroughly investigated in the context of anthrax infection since TEM8 is one of three receptors known to facilitate cellular entry of anthrax toxin.2–4 TEM8 binds specifically to protective antigen (PA), a nontoxic component of anthrax toxin. Site-directed mutagenesis analysis and x-ray crystal structure analysis indicate that, like the binding of integrins to their physiological targets, TEM8-PA binding uses high-affinity contacts to a divalent cation contained in a metal ion-dependent adhesion site (MIDAS domain).1,5–7
High similarity to integrins suggests that TEM8’s physiological role may involve angiogenic processes. Indeed, TEM8 was first described as a marker upregulated in the endothelium of human colorectal cancer compared with normal colorectal endothelium 8 and later shown to be upregulated in bladder, esophageal, and lung cancer tissues. 9 Both genetic ablation of TEM8 and anti-TEM8 antibodies can strongly attenuate tumor growth and tumor angiogenesis in mice in diverse tumor models, demonstrating a role for TEM8 expression within the host in tumor progression.9,10
TEM8 expression is increased in cultured endothelial cells during tubule formation, 11 and modulation of TEM8 expression or activity can also affect endothelial cell function in vitro. Inhibition or knockdown of TEM8 reduces migration and tubule formation,11,12 whereas overexpression increases migration. 11 Notably, TEM8 expression patterns along the vessels of human colorectal tumors mirror those for one of its ligands, collagen VI. 13 TEM8 also interacts with the extracellular matrix proteins collagen I and laminin.11,13 Treatment of endothelial cells with a recombinant TEM8 extracellular domain inhibits their binding to both gelatin and collagen I, whereas binding to other matrix proteins, fibronectin and vitronectin, is unaffected. 11 TEM8 appears to coordinate cell spreading by coordination of cell surface domain binding to external ligands (such as collagen) with cytoplasmic tail interaction with the actin cytoskeleton. 14
However, TEM8 is not simply a tether between actin and extracellular matrix proteins. TEM8 influences vascular branching and patterning in the CAM model, via an interaction with Wnt signaling cascades in endothelial cells. 15 In addition, mice with a genetic deletion of TEM8 accumulate extracellular matrix proteins in many tissues, suggesting that TEM8 may also regulate turnover of extracellular matrix proteins. 16 Furthermore, missense mutations in the TEM8 gene have been identified in individuals with infantile hemangioma, a disease characterized by localized lesions of disorganized angiogenesis. 17 More recent studies have suggested that the level of TEM8 expression within breast cancer cell lines is correlated with their capacity for growth, invasion, and metastasis, with more aggressive basal-like cells expressing higher levels of TEM8 mRNA and protein. 18 The link between TEM8 and tumorigenesis has also led to interest in manipulating the tumor-specific expression of TEM8 for the selective delivery of toxic or vascular-disrupting agents to tumor sites. 19
Together, these data show that TEM8 functions in angiogenesis and can regulate endothelial cell shape, adhesion, and migration. Importantly, we have shown that PA significantly inhibits angiogenesis in vivo. 20 This observation suggests that occupancy of TEM8 in the PA binding site could inhibit TEM8 binding to physiological ligands that mediate the receptor’s physiological impact. Here, we describe a ratiometric, steady-state, fluorescence resonance energy transfer (FRET)–based, mix-and-measure, high-throughput screening (HTS) assay that is highly effective at identifying inhibitors of the TEM8-PA interaction. The assay is based on FRET observed during the interaction of a fluorescently labeled extracellular domain of TEM8 and a fluorescently labeled PA molecule. Because extracellular matrix proteins are expected to bind TEM8 in the MIDAS domain, the same region where PA binds, the molecules identified as TEM8-PA inhibitors are also likely to inhibit TEM8 binding to ligands required for angiogenic processes and thus have potential application as both antiangiogenic and antianthrax agents.
Materials and Methods
Construction of pQE30-TEM8-mCit
A TEM8 truncation (amino acids 33–228) was amplified by PCR using the forward primer, F.BamHI.TEM8 (gatc
Protein Expression, Purification, and Labeling
PAE733C was cloned, expressed in BL21 DE3 Star Escherichia coli (Invitrogen, Carlsbad, CA), and purified using a combination of ion exchange (HP Q-Sepharose; GE Healthcare, Piscataway, NJ) and size exclusion chromatography (Sephacryl 200HR; GE Healthcare) similar to those methods previously reported. 21 Protein purity was determined to be ≥85% by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) with Coomassie staining. This single cysteine mutant was labeled with Alexa Fluor 546 C5 maleimide (Invitrogen) or QSY7 (Life Technologies, Carlsbad, CA) using manufacturer recommended methods.
TEM8-mCit, an N-terminal fusion of a monomeric EYFP variant Citrine with a TEM8 truncation of the extracellular domain, was expressed in E. coli (T7 Express; New England Biolabs, Ipswich, MA). TEM8-mCit contains an N-terminal hexahistidine tag for downstream affinity purification. Briefly, a 50-mL overnight culture was grown in ECPM1 and was used to inoculate 5 L of ECMP1 in a 5-L bioreactor. The culture was grown at 37 °C to a density of 8 to 12 OD600 and then induced with isopropyl β-D-1-thiogalactopyranoside (IPTG) at a final concentration of 0.8 mM for 3 h at 37 °C. The entire culture was harvested and centrifuged for 20 min at 5000 g. The pellet was resuspended in lysis buffer (20 mM Tris [pH 7.8], 150 mM salt, 20 mM imidazole, 0.02% Tween-20) with 4× the cell pellet volume. The resuspended cells were passed through a cell disruptor (Constant Systems, Daventry, UK), then sonicated (VWR Sonifier; VWR, Radnor, PA) 4× for 1 min each, and then passed through the cell disruptor a second time. The lysate was cleared by centrifugation at 12 000 g for 30 min. The cleared lysate was loaded onto 50 mL of nickel chelating resin (HisFF; GE Healthcare Lifesciences, Uppsala, Sweden) at 10 mL/min. Step gradients were performed at 10%, 20%, 40%, and 100% of 250 mM imidazole in lysis buffer. The fractions from 20% to 40% were pooled, concentrated by ultrafiltration (Millipore, Billerica, MA), and loaded onto a 75-mL S-200 (GE Healthcare Lifesciences) gel filtration column equilibrated in 20 mM Tris (pH 8), 150 mM salt, and 0.02% Tween-20. Fractions were analyzed by SDS-PAGE and fluorescent fractions pooled.
Prior to settling on the above method, several additional approaches for labeling TEM8 were investigated. Direct labeling of a wild-type TEM8 33–228 truncation, expressed as a glutathione S-transferase (GST) fusion in pGEX-4T-1, or identical TEM8 site-directed mutants with one or more native cysteines changed to alanines, FlAsH tagging of the TEM8 truncation with an N-terminal CCPGCC tetracysteine motif, and expression of TEM8 as a fluorescent fusion protein (described above) were all investigated. These variants of the TEM8 truncation were cloned, sequence verified, expressed in BL21 DE3 Star E. coli (Invitrogen), and purified using combinations of ion exchange (HP Q-Sepharose; GE Healthcare), affinity (GST Bind Agarose; Novagen, Madison, WI), and size exclusion chromatography (Sephacryl 200HR; GE Healthcare). Prior to downstream labeling of each expressed protein, the GST was cleaved by incubation with human α-thrombin (Enzyme Research Laboratories, South Bend, IN) as the GST was linked to the TEM8 truncation via a thrombin cleavage site. Final protein purity was determined to be ≥85% by SDS-PAGE with Coomassie staining. Single, double, or triple cysteine TEM8 mutants were labeled with Alexa Fluor 488 C5 maleimide, Alexa Fluor 546 C5 maleimide, or Alexa Fluor 647 C2 maleimide (Invitrogen) using manufacturer recommended methods. The tetracysteine-tagged TEM8 was labeled with either FlAsH or ReAsH (Invitrogen).
The dye/protein ratios of all protein conjugates was determined by UV-Vis spectrophotometry. Protein activity was assessed by using a gel-shift assay, pull-down, or fluorescence spectroscopy to measure resonance energy transfer upon PA binding TEM8 in vitro.
Validation of TEM8-PA Interaction
To test for energy transfer between TEM8-mCit and PAE733C*AF546, fluorescence spectra were acquired using a spectrofluorometer (QM-4; Photon Technology International, Birmingham, NJ) with a 75-W Xe arc lamp excitation and photon-counting photomultiplier detection. Slits for both excitation and emission monochromators were set to achieve a 4-nm bandpass. PA-alone and TEM8-Cit–alone controls were also performed. Scatter and background fluorescence was subtracted using the spectrum acquired for the PA-alone control. Fluorescence spectra of 500-nM solutions of TEM8-mCit, PAE733C*AF546, or mixtures of PAE733C*AF546 and TEM8-mCit were also acquired in HEPES buffered saline + Tween-20 (HiHBST; 50 mM HEPES [pH 7.4], 150 mM NaCl, 2 mM MgCl2, 0.1% Tween-20) following a 1-h incubation at room temperature.
A PA-TEM8-mCit binding assay consisted of mixing PAE733C labeled with biotin-PEG2-maleimide (Pierce, Rockford, IL; labeled according to manufacturer protocols) with TEM8-mCit at 1 µM each in HiHBST. The mixture was incubated for 1 h at room temperature, then with 2 µL of streptavidin functionalized magnetic beads (New England Biolabs) for an additional 15 min. Magnetic beads were pulled down with a strong magnet and subsequently washed 4 × 150 µL with HiHBST. Finally, the beads were resuspended in HiHBST and a fluorescence emission spectrum (λex = 495 nm) was acquired in a low-volume (12-µL) fused silica cuvette, using conditions described above. Control experiments were also performed in which the pull-down experiment was also performed in the absence of PA.
To measure TEM8-PA affinity using FRET, constant concentrations of TEM8-mCit (ca. 20 nM) were incubated with varying concentrations of PA-QSY7 (nM to µM) in low-volume 96 half-well plates (Corning, Corning, NY) in HiHBST with sodium azide (0.01%), at constant total volume. TEM8-mCit signal following 20- to 75-h incubation at room temperature was read using a Genios plate reader (TECAN, Männedorf, Switzerland) using 500 ± 13–nm excitation and 535 ± 13–nm emission filters. The resulting TEM8-mCit signal was corrected for the inner filter effect, 22 using estimates of the PA absorbance in each well, calculated from total PA concentration, path length, and the measured concentration dependence of PA-QSY7 absorbance at excitation (500 nm) and emission (535 nm) wavelengths. Percentage quenching of corrected emission signal was plotted versus free PA-QSY7 concentration; nonlinear regression of these data to the single-site binding isotherm yielded dissociation constant.
High-Throughput Assay Optimization
Optimization of FRET assays using the TEM8-mCit and PA-AF546 proteins was carried out in Corning 3573 384-well plates. Fluorescent proteins in HEPES buffered saline with Tween (20 mM HEPES [pH 7.4], 150 mM NaCl, 2 mM CaCl2 or 2mM MgCl2, 0.075% Tween-20) were added using a Precision Plus 2000 liquid-handling robot (Bio-Tek, Winooski, VT) in a 40-µL total volume. Parameters varied included the concentration of TEM8-mCit (250–1000 nM) and PA-AF546 (200–500 nM) used, the metal used in the HEPES buffer (either 2 mM CaCl2 or 2 mM MgCl2), and detergent concentration. There were eight replicates per variable tested. For each parameter tested, there were also eight replicate positive control wells (no interaction) that involved the presence of an excess of EDTA (5 mM) within the assay buffer. Following incubation for varied periods of time, plates were read on an EnVision (PerkinElmer, Waltham, MA) plate reader using a 485/14-nm excitation filter, with 535/25-nm and 595/60-nm emission filters (yielding Em535 and Em595, respectively) incorporating a barcode reader to correlate fluorescence measurements with plates. The FRET ratio (FR = Em595/Em535) was calculated for each well, and Z′ measurements of assay performance were calculated for each condition using FRs from eight replicate wells and the appropriate control wells (+EDTA). All FRET optimization experiments, as well as subsequent validation screens, high-throughput screens, and IC50 generating experiments, were carried out at ambient room temperature.
High-Throughput Validation
A more precise and faster liquid-handling robot (WellMate with integrated stacker, Matrix Technologies, Maumee, OH), more amenable to HTS, was used to add the assay reagents to 384-well plates for the validation assay. Specifically, 30 µL of a solution containing 333 nM TEM8-mCit and 0.125M MgCl2 in HBST (20 mM HEPES [pH 7.4], 150 mM NaCl, 0.1% Tween-20) was added to 192 wells of a barcode-labeled Corning 3573 384-well plate. An excess of EDTA (5 mM) was added to the TEM8-mCit destined for the 192 inhibitor-positive control wells to chelate metal and thereby prevent the interaction of TEM8 with PA in those wells, whereas inhibitor-negative control wells contained additional NaCl (10 mM) to provide equivalent sodium concentrations while not chelating divalent cations. Then, 10 µL of 1300 nM PAE733C*546 was added to all wells, and plates were incubated and read as above at 7-min intervals over 4 h to identify the optimal incubation time for the assay. Z′ of the FRET ratio (FR) was calculated for each time point, along with normalized difference in FRET ratios (ND = (FRneg – FREDTA)/(FRneg + FREDTA), where FREDTA is the FRET ratio of the positive control containing EDTA, and FRneg is the FRET ratio of the negative control).
High-Throughput Screen
For HTS of libraries of known bioactive molecules, 30 µL of a solution of 333 nM TEM8-mCit and 0.125M MgCl2 in HBST was added to the wells of a barcode-labeled Corning 3573 384-well plate as above. An excess of EDTA (5 mM) was added to the TEM8-mCit destined for positive control wells, to prevent the interaction of TEM8 with PA in those wells. Next, 0.3 µL of test compound (0.5–5 mg/mL or 10 mM) dissolved in DMSO was added by pin transfer using a custom Epson robot to duplicate plates. Then, 10 µL of 1300 nM PAE733C*546 was added to all wells, and plates were incubated for 1.5 h. Incubation lengths varied between individual wells, as a function of the time required for delivery of library compounds to individual positions in the well-plate. Following incubation, plates were read as above. For each plate, 32 positive control wells were generated by the addition of 5 mM EDTA to the TEM8 solution. There were also a total of 70 negative controls in which no compound was added. When F595 and F535 are plotted against each other, control wells clearly fall on two lines: one for positive and one for negative controls. However, these lines do not intersect at the origin, and thus differences in filling or optical path length will systematically and artifactually affect the FRET ratio. To minimize the effect of differences in plate filling and meniscus shape on later calculations, the FRET data were rescaled by calculating the intersection of the best-fit lines for all the positive and negative control wells in the experiment and subtracting this point from each point in the experiment, after which the FRET ratio was calculated. This corrected FRET ratio was used in further analysis.
TEM8-PA Inhibitor IC50s
Assays to identify IC50 values for TEM8 inhibitors identified from screening of known bioactive compound library plates available at the ICCB-Longwood were carried out using a protocol similar to that used in the high-throughput screens. However, these assays were conducted in a smaller volume (10 µL) in Corning 3821 low-volume 384-well plates using an EDTA-containing buffer with both interacting proteins at final concentrations similar to those used in the screening assay (250 nM TEM8-mCit, 325 nM PAE733C*AF546, 0.5 mM EDTA, in HBST). To these wells were added 100 nL of serial dilutions of cisplatin, ebselen, celastrol, phenylmercuric acetate, or thimerosal in DMSO. Following compound addition, 500 nL of 20 mM MgCl2 was added to activate the assay. Positive controls (24 wells) for inhibition of the TEM8-PA interaction were generated by omitting Mg2+. Plates were incubated for 1.5 h and read as above. DMSO was added to 24 negative (for inhibition of interaction) control wells instead of compound. The F595/F535 fluorescence emission ratio was measured and plotted as a function of final inhibitor concentration. Data were fit to a single-site binding isotherm using GraphPad Prism 5 (GraphPad Software, La Jolla, CA). Identical assays were carried out in the presence of 50 µM reduced glutathione (Sigma, St. Louis, MO), to determine its effect on the ability of the identified TEM8 inhibitors to inhibit the TEM8-PA interaction.
Results
FRET Screening Assay Design
FRET is the highly distance-dependent transfer of energy from a donor molecule to an acceptor molecule. For FRET to occur between two molecules, they must be in sufficient proximity (usually less than 100 Å), and the donor’s emission spectrum must overlap with the acceptor’s excitation spectrum. In this case, excitation of the donor molecule will lead to a quenched donor emission spectrum and an enhanced acceptor emission, as the energy from the donor molecule excites the acceptor molecule. Because of the length scale and distance dependence, FRET is a sensitive probe of macromolecular association and can therefore be employed to study protein-protein binding. We have previously used a high-throughput FRET assay to identify inhibitors of CMG2. 23 Using a recombinant TEM8 fusion with a fluorescent protein and directly labeled PA in a simple homogeneous assay format, we have developed a robust high-throughput FRET screening assay to identify small molecules that inhibit the PA-TEM8 interaction.
The development of this assay, like all other FRET assays, required production of two fluorescently labeled proteins with overlapping spectra. We first attempted to directly label our protein reagents with small-molecule fluorescent dyes following expression as GST fusion proteins, in a similar manner to the approach previously used in the development of a high-throughput CMG2-PA FRET assay. 23 We could successfully label the PA cysteine mutant with either Alexa Fluor 488 or Alexa Fluor 546. Dye/protein conjugate ratios were between 0.85 and 1.15 for PA. However, efforts to label TEM8 with small-molecule dyes were less successful. We attempted to directly label four different TEM8 proteins: wild-type and variants with one, two, or all native cysteines mutated. TEM8 proteins in which one or more cysteines were mutated were only partially soluble after cleavage of GST, and these proteins did not bind PA in gel-shift assays or compete with CMG2 for PA binding ( Table 1 ). Not surprisingly, none of these directly labeled TEM8 proteins exhibited FRET when incubated with fluorescently tagged PA. As a competing strategy to label TEM8 with small-molecule dye(s), we appended a FlAsH-reactive tag onto the TEM8-GST fusion protein. With the FlAsH tag (i.e., TEM8-CCPGCC-AF647), the TEM8-GST fusion protein was both active in the gel-shift assay and soluble. When the labeled TEM8 was incubated with PA-AF566, significant FRET was observed, whereas incubation with PA-AF546 produced minimal FRET. As an alternative labeling strategy, we turned to expressing TEM8 as a fusion protein with an EYFP variant (mCit), using a His-tag for purification. The TEM8-mCit-His fusion protein was well expressed, purified well, and exhibited PA binding in a pull-down with biotinylated PA and streptavidin magnetic beads ( Fig. 1A ). In addition, we observed significant FRET when the fusion protein was incubated with PA-AF546 ( Fig. 1B ). A clear decrease in intensity of the donor (TEM8-mCit) at ~525 nm (donor quenching) and some enhancement of the intensity of the acceptor (PAE733C*546) at ~572 nm (sensitized emission) was observed, as compared with controls with donor (TEM8-mCit) alone ( Fig. 1B ). Given the greater simplicity in expression and reduced production cost for TEM8-mCit relative to CCPGCC-AF647, we selected the TEM8-mCit-His fusion protein to develop the HTS assay. Using this protein pair, FRET is observed as decreased fluorescent emission at 525 nm (from donor molecule TEM8-mCit) and increased fluorescent emission signal at 595 nm (from the acceptor molecule PA-AF546) after direct donor excitation at 488 nm. The utility of FRET measurements for observation of the TEM8-PA interaction is further supported by FRET affinity measurements of TEM8-mCit and PA ( Fig. 1C ); we observe good agreement between Kd values measured by FRET (108 ± 14 nM [± 95% confidence limit]) and the previously published value of 130 nM obtained using an orthogonal assay. 24
Outline of Methods Attempted for Generation of a TEM8-PA FRET Assay and Their Results.

AF546 = AF546-C5-maleimide. AF647 = AF647-C2-maleimide. *C1 = TEM8 C178A C221A. †C2 = TEM8 C39A C221A. ‡C3 = TEM8 C39A C178A. **TEM8-CCPGCC-AF647. FRET, fluorescence resonance energy transfer; N, no; NA, not applicable; PA, protective antigen; TEM8, tumor marker endothelial 8; Y, yes.
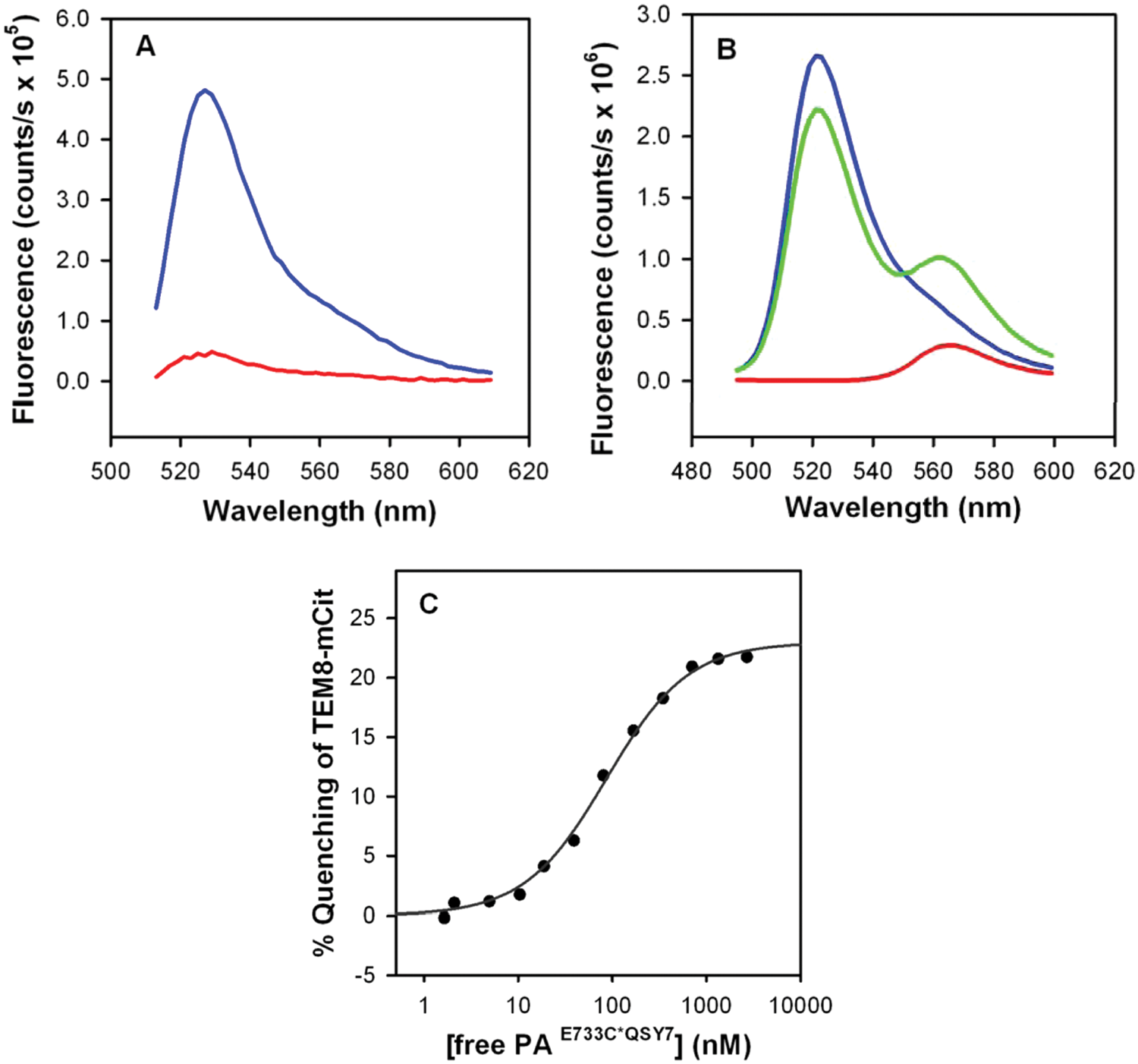
Binding of tumor marker endothelial 8 (TEM8)–mCit to protective antigen (PA). (
The FRET behavior of the TEM8-mCit/PA AF546 pair became the basis of our screening assay. Small molecules that inhibit the interaction of these two molecules reduce the observed FRET signal observed in our assay. Specifically, inhibition of the interaction results in an increased fluorescent emission from the donor molecule (TEM8-mCit; λmax 525 nm) and a reduced fluorescent emission signal from the acceptor molecule (PA-AF546 λmax 572 nm), after direct excitation of mCit at 488 nm. Our assay measures the FRET ratio for TEM8-mCit and PA-AF546 in the presence of possible inhibitors and compares that value with the FRET ratio observed for labeled TEM8 and PA in the absence of inhibitor, under binding conditions. Significant reductions in the observed FRET ratio are then used to infer inhibition of the TEM8-PA interaction by individual library compounds.
High-Throughput Screen Optimization and Validation
To translate the FRET assay outlined above to a sensitive high-throughput format, it was necessary to adjust experimental conditions to optimize the assay performance. Screen performance reflects a series of different parameters, including stability, sensitivity, reproducibility, robustness, and well-to-well variation across the screening plate. A measure of screen performance can be provided by the observed Z′ value, which is calculated from the mean and standard deviation of the positive and negative control wells. 25
In this case, the positive control represents inhibition of the TEM8-PA interaction (and loss of observed FRET), whereas the negative control represents no disruption of TEM8-PA binding, with corresponding FRET. A divalent metal cation is required for the interaction of TEM8 with PA. 2 Hence, a positive control can be generated either by omission of the metal ion or addition of excess (5 mM) EDTA, whereas the negative control (i.e., no inhibition of binding) is the observed FRET ratio in the presence of divalent metal. Calculation of the mean and standard deviation of the observed FRET ratios in both positive and negative control wells then yields Z′. Generally, a Z′ value of >0.5 is the minimum acceptable value for an interpretable screening assay, whereas a Z′ value of >0.7 is considered indicative of a robust and highly reproducible assay.
Having demonstrated the stability of both labeled PA and TEM8, and having observed FRET resulting from TEM8-mCit binding to PA-AF546, we searched for assay conditions that would result in high observed Z′ values. Attempts were made to optimize the incubation time, choice of divalent metal (Ca2+ vs. Mg2+), and the PA:TEM8 ratio. Initial experiments used a constant concentration of TEM8-mCit (500 nM) and different concentrations of PA-AF546 (250–1000 nM), using conditions similar to those previously used for a CMG2 FRET assay developed in our laboratory. 23 EDTA was added to positive control wells to abolish TEM8-PA binding via sequestration of solution Ca2+ required for binding. No inhibitor compounds were added during these model experiments, as Z′ is calculated from control wells alone. Results of these experiments (not shown) demonstrated that in positive control (EDTA-containing) wells, the emission at 535 nm (TEM8 donor signal) remained constant, whereas emission at 595 nm (PA acceptor signal) increased, with increasing AF-546 concentration. This behavior is consistent with lack of the TEM8-PA interaction and resulting lack of FRET in positive control wells. In contrast, negative control wells (no EDTA) showed decreases in TEM8 fluorophore signal (donor quenching) at 535 nm as a function of increasing PA concentration, as well as increases in PA fluorophore signal (595 nm) larger than those expected based on increased PA concentration alone (sensitized emission). These observations indicate that energy was transferred from mCit-TEM8 to AF546, consistent with a TEM8-PA interaction in negative control wells. Fluorescent emission at 535 nm (donor emission) and 595 nm (acceptor emission) in positive and negative control wells was measured every 5 min over a 12-h period. The Z′ for these experiments rose rapidly over the first hour, settling at a value ranging from ~0.4 to 0.6 (data not shown). The rise in Z′ over time coincided with an increase in emission at 595 nm and a decrease in emission at 535 nm only in the negative control wells, indicating that it resulted from increased FRET (presumably due to increased TEM8-PA association over time).
We next compared assay performance when Mg2+ and Ca2+ were used as the metal ion. We observed much improved Z′ values and much more rapid apparent association when Mg2+ served as the metal ion (
An alternative way to present data from these FRET screens is in scatterplot format, in which the donor (x-axis) and acceptor (y-axis) fluorescence intensities are plotted for individual wells, as shown in Figure 2B . In this format, the slope of the line connecting a point with the origin is the FRET ratio, which is largely affected by protein-protein interaction. The distance along that line is an indication of overall fluorescence intensity, which can be affected by such factors as differences in path length, extent of filling, and meniscus shape. In Figure 2B , two well-separated tight clusters of data are observed: one for the positive control, and one for the negative control. Data for positive inhibition control wells are characterized by a reduced FRET, reflected in a position below and to the right (i.e., lower acceptor fluorescence and higher donor fluorescence), versus negative control wells. Figure 2B demonstrates clear separation of positive and negative controls, indicating that binding and inhibition are extremely well separated in our screen. Since inhibitors are identified by their relative proximity to positive controls in the screening scatterplots, good separation between positive and negative controls, as seen in our assay, promotes highly sensitive screening.
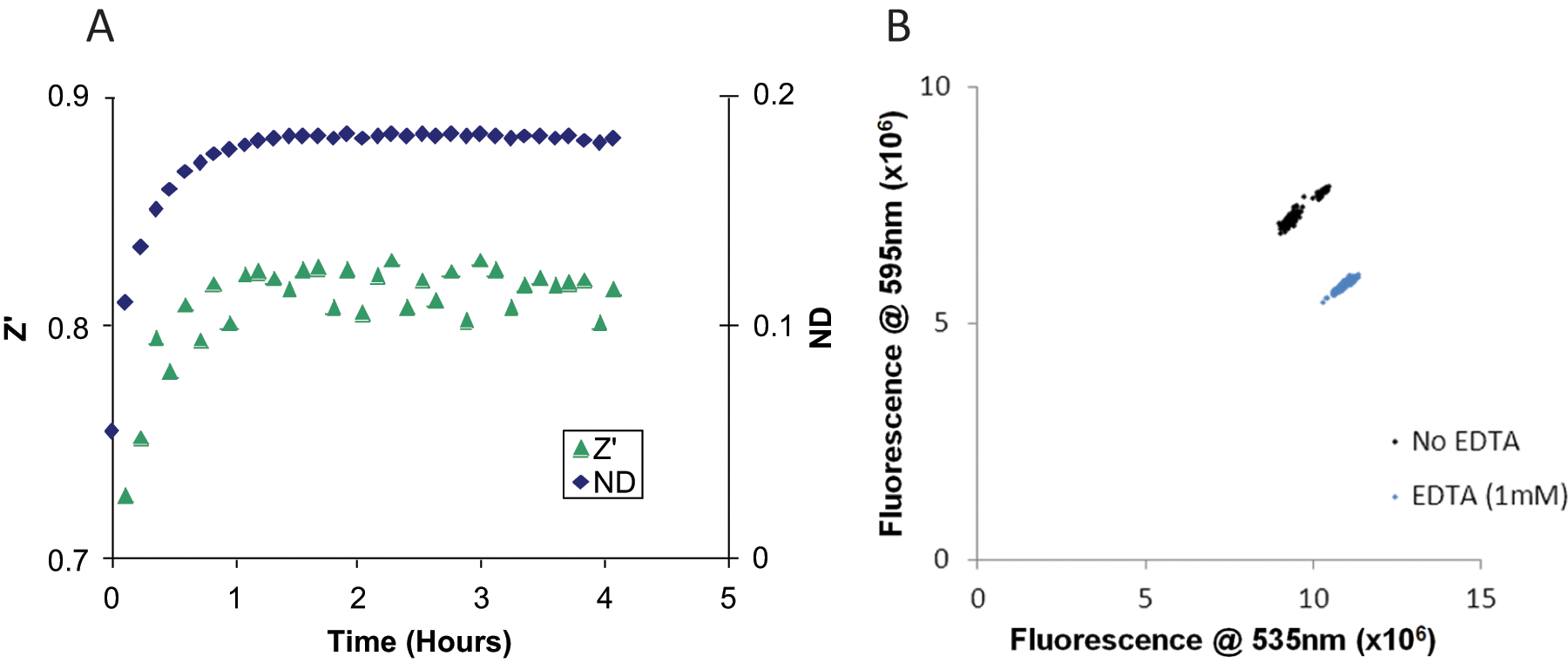
Assay validation. Using the conditions described, 192 wells of standard assay conditions and 192 wells of standard assay conditions + 10 mM EDTA were prepared, excited at 485 nm, and read at 535 and 595 nm. (
Pilot Screen Identifies Inhibitors of the TEM8-PA Interaction
Once assay behavior was validated and optimized for positive and negative controls, we conducted pilot screens of a library of 2310 known bioactive small molecules. The library contained compounds with high structural diversity, collective coverage of multiple therapeutic areas, and known safety and bioavailability in humans. For example, the range of compounds includes ion channel blockers, G-protein–coupled receptor (GPCR) ligands, second-messenger modulators, nuclear receptor ligands, actin and tubulin ligands, kinase inhibitors, protease inhibitors, gene regulation agents, lipid biosynthesis inhibitors, and other well-characterized compounds that perturb cell pathways, including a significant subset of substances known to influence brain activity. Approximately 50% of Food and Drug Administration (FDA)–approved drugs were included in the library.
Screening assays were conducted in 384-well plates, with fluorescent readout at 535 nm and 595 nm for each well. Each plate contained negative controls (Mg2+ present, no compound added) and positive controls (absence of Mg2+, no compound added). Each compound was assayed on two duplicate plates, to minimize use of library compounds while minimizing the possibility of false negatives. To visualize the screen results, fluorescence data for each well, including positive and negative controls, was plotted in scatter format (donor fluorescence at 535 nm vs. acceptor fluorescence at 595 nm) ( Fig. 3 ). Inhibitory “hit” compounds are those that lie between the positive and negative controls; proximity to positive controls is symptomatic of greater inhibition. Wells with acceptor or donor fluorescence outside the range of control wells are assumed to reflect strongly fluorescent and/or strongly absorbent compounds whose altered FRET ratio(s) may not reflect binding inhibition.
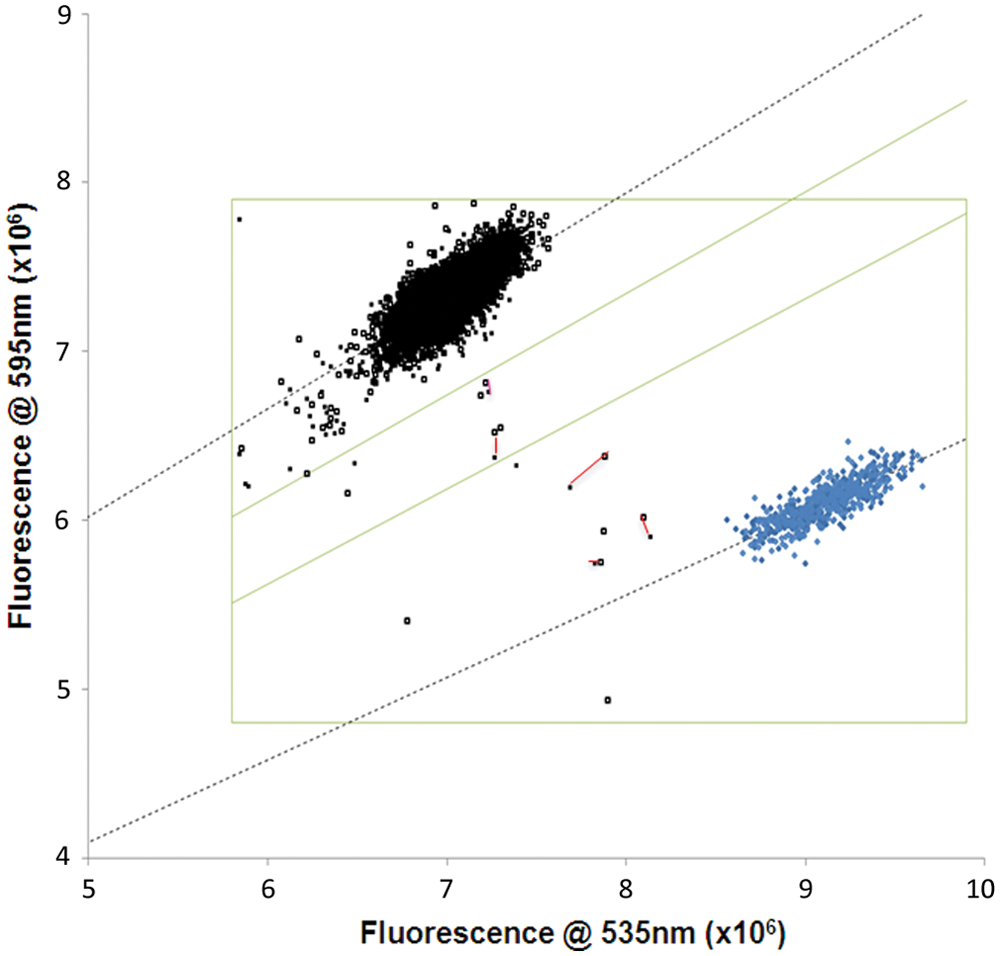
Pilot study for inhibitors of the protective antigen (PA)–tumor marker endothelial 8 (TEM8) interaction. Ten library plates containing 2310 known bioactive compounds (black, upper left) and 640 EDTA-containing positive control wells (blue, lower right) were assayed by fluorescence resonance energy transfer (FRET). Scatterplot of donor (Em535) and acceptor (Em595) emission upon excitation at 485 nm. The green box identifies wells unaffected by compound fluorescence or absorbance, while the diagonals demarcate the regions of 25% and 50% inhibition. Duplicate wells for inhibitors are connected by red lines. They are, from upper left to lower right, thimerosal, phenylmercuric acetate, cisplatin, and ebselen (2 pairs).
To quantify the strength of “hit” compounds in the screen, the average FRET ratio (calculated from two plates) was calculated for each compound and compared with the average FRET ratios from both the negative and positive control wells. In this case, a percentage shift from the negative controls (FRET, TEM8-PA interaction) toward the positive controls (EDTA, no interaction) was then calculated for each compound well. A “strong hit” was assigned to compounds in which both evaluated wells inhibited by more than 50%, a “medium hit” was assigned to compounds that inhibited by 25% to 50%, and a “weak hit” was assigned to those compounds in which only one evaluated well inhibited by >25%. Using these criteria to evaluate the screening data shown in
Figure 4
, we identified four strong hits, representing three different compounds: ebselen (measured twice), cisplatin, and gentian violet. The dramatic decrease in emission at both 535 nm and 595 nm in the gentian violet samples indicated that this “hit” is an artifact resulting from light absorption by this highly colored compound; it was excluded from further study. We have previously identified cisplatin as an inhibitor of CMG2-PA interactions
23
and carried out analysis indicating that cisplatin nonspecifically alkylated both PA and CMG2; a similar mechanism is likely at work for the TEM8-PA interaction. Medium hits were thimerosal, phenylmercuric acetate, and celastrol, with the latter hit also resulting from compound absorbance (see below). The structures of the compounds identified as strong or medium hits are shown in
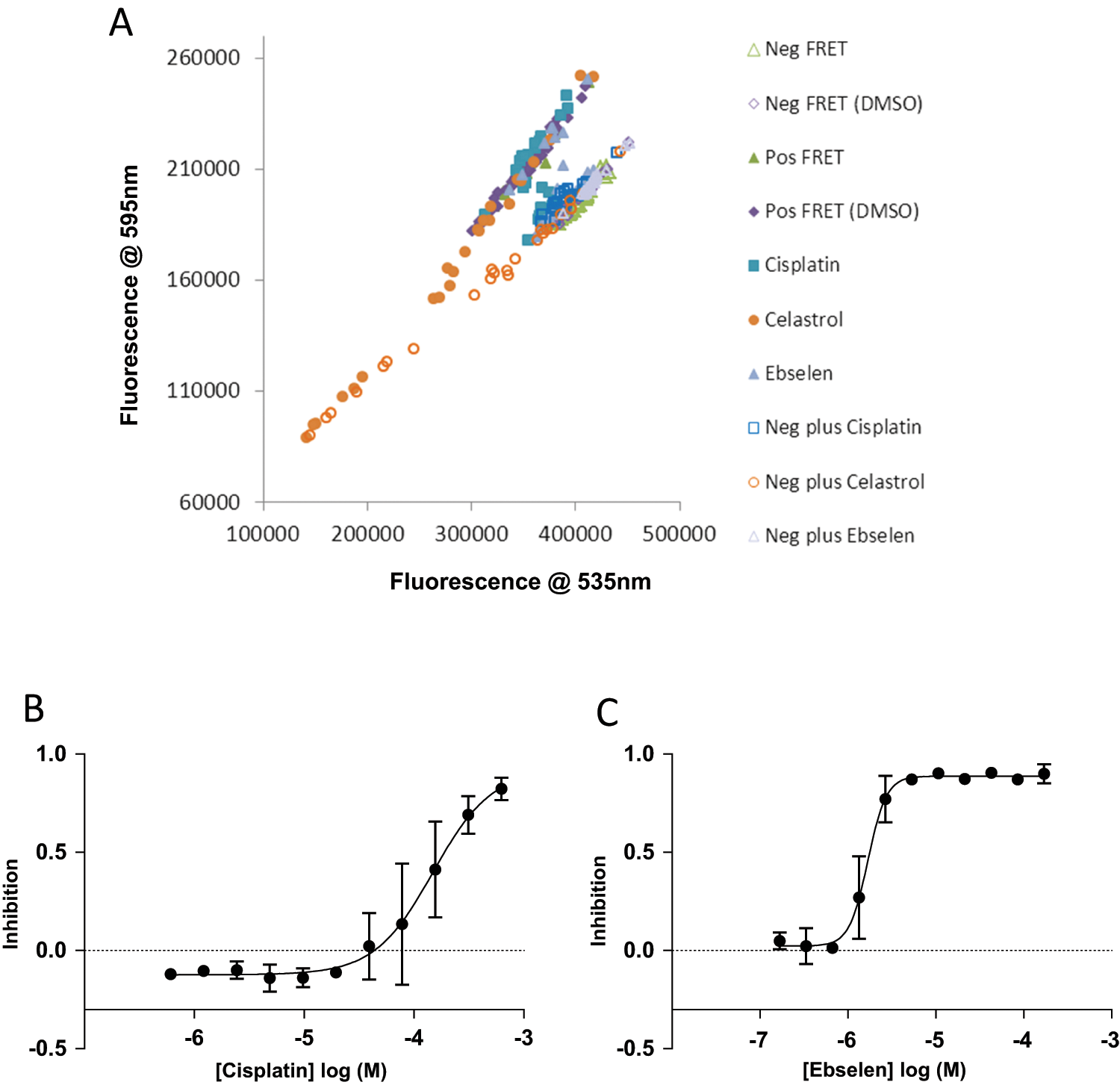
IC50 of cisplatin and ebselen for protective antigen (PA)–tumor marker endothelial 8 (TEM8) interaction. TEM8 inhibitors identified from the initial fluorescence resonance energy transfer (FRET) library screen were tested again in duplicate at 11 different concentrations to generate a concentration curve and an IC50 value. (
Secondary Characterization of Identified Inhibitors
Experiments aimed at calculating IC50 values for cisplatin, ebselen, and celastrol, thimerosal, and phenylmercuric acetate were carried out, and where greater than 50% inhibition was achieved, IC50 values were generated from F595-nm/F535-nm fluorescence emission ratios, normalized to the positive (DMSO vehicle control only) and negative control (no Mg2+) ratios, for each concentration of compound tested. Celastrol was a compound of bright orange color and was determined to be an absorbing compound ( Fig. 4A ). Thus, an IC50 could not be generated. Its original classification as a hit appears to have resulted from a concentration on the original screening plate sufficiently low that it was not flagged in the original screen as absorbing. Both cisplatin and ebselen inhibited FRET with IC50 values of 147.7 µM and 1.7 µM, respectively ( Fig. 4B,C ). The Hill coefficient for cisplatin was 1.6, whereas the Hill coefficient for ebselen was 4.1. We have previously reported that cisplatin inhibits both endothelial cell migration and angiogenesis in the corneal micropocket assay. 23 However, given the well-documented effects of cisplatin on DNA, we do not believe that these effects are specific to interaction with the anthrax toxin receptors. Phenylmercuric acetate and thimerosal were “medium hits” in the original screen. Assays aimed at determining IC50s for these compounds were also conducted and dose-dependent inhibition was observed, but 50% inhibition of binding did not occur at concentrations up to 1 mM (data not shown).
Having noted that each of the identified inhibitors is thiol reactive, we sought to determine whether the addition of excess thiol might protect TEM8 from inhibition. The inhibitory activity of ebselen, cisplatin, phenylmercuric acetate, and thimerosal was assessed in the presence and absence of 50 µM glutathione. In each case, there was a significant decrease in inhibition of binding when glutathione was present, with the IC50 increasing 7-fold and 250-fold for cisplatin and ebselen, respectively (
Discussion
We have described a sensitive and robust high-throughput assay that identifies inhibitors of the interaction of TEM8 with one of its known ligands, protective antigen. Consequently, molecules identified from this pilot screen and from larger screens of small molecules have the potential to be developed as therapies for anthrax infection as well as tumor growth and other angiogenic diseases. The assay has great potential to identify novel small-molecule inhibitors of TEM8, which can subsequently be tested for antitumor and antianthrax applications. The assay is also suitable for screening antibodies for TEM8 inhibitory activity; we have been able to demonstrate inhibition with one such antibody (data not shown).
As outlined in the Introduction, TEM8 also represents an attractive target for adjuvant antiangiogenic therapy for solid tumors. Importantly, it is possible that TEM8 inhibition may result in little “on-target” toxicities because TEM8 knockout mice display no significant serious physiological abnormalities. The exception to this is misalignment of incisor teeth due to accumulation of extracellular matrix proteins and altered female fertility resulting from essential nonredundant TEM8 function in the uterus. 10 TEM8 levels are elevated on tumor endothelium compared with quiescent endothelium, and therapies targeting TEM8 appear to be effective for tumor angiogenesis without affecting angiogenesis in other tissues.9,26 However, further work to determine expression levels and function of TEM8 in normal human tissues is warranted as TEM8 inhibitor therapeutics are developed.
Obviously, inhibition of TEM8 can also be an effective strategy for amelioration of anthrax toxicity, either alone or combined with CMG2 inhibitors. Although studies in knockout mice indicate that CMG2 rather than TEM8 plays a central role in allowing the entry of anthrax toxin into macrophages and causing lethal effects, therapeutic molecules that target TEM8 rather than CMG2 have proved more effective in rat models. 27 Hence, small molecules identified in our high-throughput screens as inhibitors of TEM8 may be further developed for application in the treatment of anthrax infection.
Our pilot screen identified several compounds that inhibited the TEM8-PA interaction and are therefore likely to inhibit TEM8 binding to physiological ligands. Interestingly, many of the molecules identified in the current screen contain a cysteine-reactive thiol metal. For example, ebselen, a selenium-containing compound, is known to modify cysteine residues of papain and other proteins.28–31 Taken in the context of our discovery that alteration of one or more cysteines in TEM8 affected both solubility of the protein and also its ability to bind to PA, this observation highlights the importance of the cysteine residues for TEM8 binding to PA and may also suggest that some families of cysteine interacting molecules may represent candidate TEM8 inhibitors. Ebselen strongly inhibited the interaction of TEM8 with PA with an IC50 of 1.7 µM. However, the high Hill coefficient value (4.1) for this compound indicates that inhibition does not follow a simple model and that cooperativity may be involved. A model whereby ebselen inhibits TEM8 interactions by reacting with any of the three cysteines on the TEM8 protein would rationalize this Hill coefficient. The ability of glutathione, a cysteine-containing molecule, to reduce the efficacy of lower concentrations of ebselen to inhibit the TEM8-PA interaction provides further support for this model. It remains to be seen if molecules that can inhibit TEM8 by modifying cysteine residues in our high-throughput assay have promise as inhibitors of TEM8 in cell culture or in animals. Experiments are ongoing in our laboratory to address that question. Existing data are encouraging. For example, the inhibitory activity of ebselen on cysteine residues within the heme enzyme indoleamine 2,3-dioxygenase in solution is replicated in macrophages in culture. 30
However, ebselen appears to be a somewhat promiscuous hit. A recent review of PubChem reveals that ebselen was found to be active in 193 of 1023 screens, with 126 of these hits coming from confirmatory screens. The known ability of ebselen to modify cysteine residues may be at least partly responsible for its promiscuity.28–31 However, despite the high activity of ebselen in high-throughput screens, it is an investigational drug for a number of different conditions, and thus far no safety concerns have been reported. Due to its antioxidant properties, it was investigated in the setting of stroke and shown to improve outcome if given within 24 hours of onset. 32 Ebselen is currently under investigation for the treatment and prevention of hearing loss (SPI-1005; Sound Pharmaceuticals, Seattle, WA) and for the treatment of vascular dysfunction in diabetes mellitus (Brigham and Women’s Hospital, Boston, MA).
This assay identifies compounds that inhibit the TEM8-PA interaction, rather than compounds that interact with TEM8. As a result, the assay could isolate compounds that interact with PA. Although such compounds could have application as antianthrax agents, they would not have utility as antiangiogenic therapies. We have previously applied the compound libraries used here to identify inhibitors of the CMG2-PA interaction, using a similar FRET assay. A comparison of results of these two pilot screens is illuminating, as compounds that interact with PA rather than TEM8 should be identified in both assays. Despite the high structural and sequence similarity between TEM8 and CMG2, there is very little overlap between inhibitors identified in the two screens. Only one compound, cisplatin, was identified in the strong and medium hits of both assays. Our previous analysis suggests that cisplatin inhibits the CMG2-PA interaction by nonspecific alkylation of both CMG2 and PA 23 ; a similar process likely also occurs in the TEM8-PA assay. Given the relatively low number of inhibitory compounds identified in both assays, it is likely that FRET inhibitors identified in the current screen bind TEM8, rather than PA. Our ongoing screens of larger numbers of compound plates containing uncharacterized chemical compounds for inhibition in both the CMG2-PA and TEM8-PA assays also indicate low overlap between the two assays (data not shown). Hence, we conclude that compounds isolated from the current screens exert inhibition by binding TEM8, rather than PA.
In summary, we have outlined a robust high-throughput assay that can identify inhibitors of TEM8. We anticipate that screening larger compound libraries to identify TEM8 inhibitors will result in the identification of potent lead molecules that can then be developed as investigational new drugs. Applications for TEM8 inhibitors may include treatment of tumor growth due to antiangiogenic mechanisms and direct effects on tumor cells, antiangiogenic treatment of other diseases such as age-related macular degeneration, and inhibition of anthrax toxin.
Footnotes
Acknowledgements
The authors thank the ICCB-Longwood screening facility for access to its libraries, equipment, screening supplies, and technical advice.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health 1R03NS053690-01 (K.A.C.) and 1R01EY018829-01 (M.S.R.), the Department of Defense W81XWH-08-1-0710 (M.S.R.), and the Susan G. Komen Foundation KG101356 (L.M.C. fellowship).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.