
Abstract
To provide a high-throughput screening method for human ether-a-go-go-gene–related gene (hERG) K+ channel inhibition, a new recombinant cell line, in which single action potential (AP)–induced cell death was produced by gene transfection. Mutated human cardiac Na+ channel Nav1.5 (IFM/Q3), which shows extremely slow inactivation, and wild-type inward rectifier K+ channel, Kir2.1, were stably co-expressed in HEK293 cells (IFM/Q3+Kir2.1). In IFM/Q3+Kir2.1, application of single electrical stimulation (ES) elicited a long AP lasting more than 30 s and led cells to die by more than 70%, whereas HEK293 co-transfected with wild-type Nav1.5 and Kir2.1 fully survived. The additional expression of hERG K+ channels in IFM/Q3+Kir2.1 shortened the duration of evoked AP and thereby markedly reduced the cell death. The treatment of the cells with hERG channel inhibitors such as nifekalant, E-4031, cisapride, terfenadine, and verapamil, recovered the prolonged AP and dose-dependently facilitated cell death upon ES. The EC50 values to induce the cell death were 3 µM, 19 nM, 17 nM, 74 nM, and 3 µM, respectively, whereas 10 µM nifedipine did not induce cell death. Results indicate the high utility of this cell system for hERG K+ channel safety assay.
Introduction
In cardiac ventricular myocytes, human ether-a-go-go-gene-related gene (hERG; KCNH2) is a critical ion channel to form early repolarization phase of action potential (AP) as a rapidly activating delayed rectifier K+ current (IKr). Loss-of-function mutation of hERG leads to potentially lethal inherited long QT syndrome.1,2 The long QT interval is also occasionally elicited by medication due to the block of the hERG K+ channel, and it can be a fatal side effect known as torsades de pointes. 3 Although hERG channel blockers such as nifekalant are useful as antiarrhythmic drugs under some particular settings, more than 50 drugs commercially available as therapeutic agents in noncardiovascular categories have been reported to elicit long QT. 4
Based on the recommendation from the regulatory authorities, any potential drug candidates in the developing stages are assessed for the activity to prolong the QT interval. Accordingly, several methods in vitro are now available to evaluate the potential of drugs to suppress hERG or prolong the QT interval. The high-throughput screening (HTS) system for the hERG inhibition assay, including Rb+ flux measurement, 5 potential sensitive fluorescence assay, 6 and improved automated patch clamp system, 7 , 8 have been also developed. New methods are, however, still emerging to evaluate the potential of test compounds to block the hERG K+ channel as early as possible in the drug-development process with lower cost, smaller effort, and no exaggerated equipment.
The present study was undertaken to develop a novel screening system to evaluate hERG channel inhibition by a simple and popular assay of cell death, such as the MTT method. We produced a novel cell system in which mutated Na+ (Nav1.5) channels and wild-type inward rectifier K+ channel (Kir2.1) and hERG K+ channels are co-expressed. When the hERG channel was suppressed by a test compound, cell death due to occurrence of a prolonged AP upon single electrical stimulation (ES) can be easily measured by MTT assay. This screening system fits well to the true HTS for hERG inhibition assay.
Materials and Methods
Cell preparation
In accordance with a previous study, 9 we mutated the triplet IFM in the III–IV interdomain linker to QQQ in the human cardiac Nav1.5 channel (Genbank: NM_198056.2). To construct the site-directed mutants of Nav1.5, Ile1485, Phe1486, and Met1487 were substituted for glutamine by a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Nav1.5 and mutated Nav1.5 (IFM/Q3) were ligated into mammalian expression vectors pcDNA3.1(+)/Neo (Invitrogen, Carlsbad, CA), respectively, using the TaKaRa ligation kit version 1 (TaKaRa, Osaka, Japan). 10 Human Kir2.1 (Genbank: NM_000891.2) was ligated into mammalian expression vectors pcDNA3.1(+)/Zeo (Invitrogen). HEK293 cells were maintained at 37 °C in 5% CO2 with high-glucose Dulbecco’s modified Eagle’s medium (Wako, Osaka, Japan) containing 10% fetal bovine serum (Invitrogen), 100 U/mL penicillin (Wako), and 0.1 mg/mL streptomycin (Meiji Seika, Tokyo, Japan). Stable expression of Kir2.1 was achieved by using calcium phosphate co-precipitation transfection techniques. After transfection, cells were cultivated for approximately 1 mo in the presence of Zeocin, and a single cell resistant to Zeocin was selected as a clone expressing Kir2.1. Double stable expressions of IFM/Q3 and Kir2.1 were done by the same method using cells stably expressing Kir2.1. G418/Zeocin-resistant cells were selected as those that were IFM/Q3 and Kir2.1 co-expressing. Double stable cells of IFM/Q3 and Kir2.1 were transiently co-transfected with hERG (Genbank: NM_000238.2) and a green fluorescent protein (GFP) by the Lipofectamine 2000 (Invitrogen). Similarly, a stable cell of Kir2.1 was transiently co-transfected with Nav1.5. GFP-positive cells were used for patch-clamp recordings from 24 to 48 h after transfection.
Solutions
The standard HEPES-buffered solution for electrophysiological recording and image analyses had an ionic composition of 137 mM NaCl, 5.9 mM KCl, 2.2 mM CaCl2, 1.2 mM MgCl2, 14 mM glucose, and 10 mM HEPES. The pH of the solution was adjusted to 7.4 with NaOH. The pipette solution for whole-cell recordings contained 140 mM KCl, 4 mM MgCl2, 10 mM HEPES, 2 mM Na2ATP, and 0.05 mM EGTA. The pH of the pipette solution was adjusted to 7.2 with KOH. The 140 mM K+ HEPES-buffered solution for DiBAC4(3) fluorescence imaging had an ionic composition of 2.9 mM NaCl, 140 mM KCl, 2.2 mM CaCl2, 1.2 mM MgCl2, 14 mM glucose, and 10 mM HEPES. The pH of the solution was adjusted to 7.4 with NaOH. Phosphate-buffered saline had an ionic composition of 137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, and 1.5 mM KH2PO4.
Electrophysiological Experiments
The whole-cell patch clamps were applied to single cells using the CEZ-2400 amplifier (Nihon Kohden, Tokyo, Japan), EPC-7 amplifier (List Electronics, Darmstadt, Germany), and data acquisition/analysis system (Digidata 1440A and pClamp software version 10; Molecular Devices, Sunnyvale, CA). A small tip of cover glass, on which cells were cultured, was placed at the bottom of a recording chamber of 500 µL in volume on the stage of the inverted microscope (TE300; Nikon, Tokyo, Japan). Cells were continuously perfused with an extracellular solution. Whole-cell recordings were carried out at room temperature.
Membrane Potential Measurements by Voltage-Sensitive Fluorescent Dye
In some experiments, membrane potentials were measured by voltage-sensitive fluorescent dye, DiBAC4(3) (DOJINDO, Kumamoto, Japan), with excitation wavelength at approximately 488 nm and emission at 512 nm, respectively. 10 Data were collected and analyzed using the ARGUS/HiSCA imaging system (Hamamatsu Photonics, Hamamatsu, Japan). ES was performed through paired platinum wire electrodes placed in the recording chamber by applying square pulses of 200 ms in duration and 200 mA in amplitude, every 1 s three times.
MTT Assay
Cell viability was monitored by the colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma-Aldrich, St. Louis, MO) assay as described previously.11,12 MTT was dissolved in phosphate-buffered saline at 5 mg/mL and filtered to sterilize and remove a small amount of insoluble residue present in some batches of MTT. Cells were seeded onto 96-well plates (approximately 5000 cells per well) and cultured for 1 day. To apply ES, a pair of platinum stimulating electrodes was inserted into each well, and the tips of electrodes were immersed in the cultivation solution. ES was performed by applying single 200 ms pulse every 1 to 3 min three times with an adjustable electrical stimulator (SEN-3301; Nihon Kohden, Tokyo, Japan). After ES, cells were incubated for 1 day at 37 °C in 5% CO2, and stock MTT solution (10 µL per 100 µL medium) was added to all wells of an assay plate, and the plates were incubated at 37 °C for 4 h. After 4 h, 20% w/v SDS (50% N,N-dimethyl formamide and demineralized water) was added to all wells, and the plates were incubated at 37 °C for 6 to 8 h. These plates were then analyzed following absorption on a Multiscan JX (Ver1.1 system; Thermo Labsystems), using a test wavelength of 595 nm and a reference wavelength of 650 nm. Unless specified in the text, no mixing was performed, and no medium was removed prior to the addition of any ingredient.
Trypan Blue Exclusion
Cell viability was determined also by the measurement of the capacity to exclude the vital dye trypan blue. ES was performed in the same manner as membrane potential measurements with DiBAC4(3). After the ES, cells were incubated for 5 min, 15 min, 30 min, and 1 h at room temperature and then stained with phosphate-buffered saline containing 0.15% trypan blue (Sigma-Aldrich) for 5 min. Blue-stained and unstained cells were counted in the viewing field of the microscope to calculate the percentage of nonviable cells.
Chemicals
Lidocaine, nifedipine, cisapride, terfenadine, and verapamil were obtained from Sigma-Aldrich. Nifekalant and barium chloride were obtained from Wako Pure Chemicals. Quinidine was obtained from Tokyo Chemical Industry (Tokyo, Japan). E-4031 was a gift from Eisai (Tokyo, Japan). The test compounds were dissolved with dimethyl sulfoxide or distilled water.
Statistics
Data are expressed as means ± SEM. The statistical significance between the two groups and among multiple groups was evaluated using Student’s t-test and Scheffe’s test after F-test or one-way analysis of variance, respectively. The Z′ factor was calculated from each dose response set as follows: Z′ factor = 1 – 3(SD of positive controls + SD of negative controls)/(mean of positive controls – mean of negative controls). 13
Results
Production of a Cell Line Expressing Mutated Nav1.5 Channel
First, we aimed to produce a recombinant cell line that elicits an extremely long AP by transfecting HEK293 cells with a mutated voltage-dependent Na+ channel, Nav1.5. A prolonged AP due to slow inactivation of mutated Nav1.5 may lead the cell to death. The replacement of the three amino acids (Ile1485, Phe1486, and Met1487) with glutamine (IFM/Q3) completely removes fast inactivation. 14 This mutation, however, killed cells presumably because of sustained Na+ influx through the noninactivating Na+ channel. It has been also known that co-expression with IFM/Q3 of the inward-rectifier K+ channel Kir2.1, which provides more negative resting membrane potential, is effective to prevent the cell death. 15 Based on these lines of information, a cell line stably expressing both Kir2.1 and IFM/Q3 was generated.
In HEK293 cells stably co-expressing Kir2.1 and wild-type or mutant Nav1.5 channels (WT+Kir and IFM/Q3+Kir), membrane currents were measured using whole-cell patch-clamp recordings (
Fig. 1A
,
B
; see also I-V relationships in
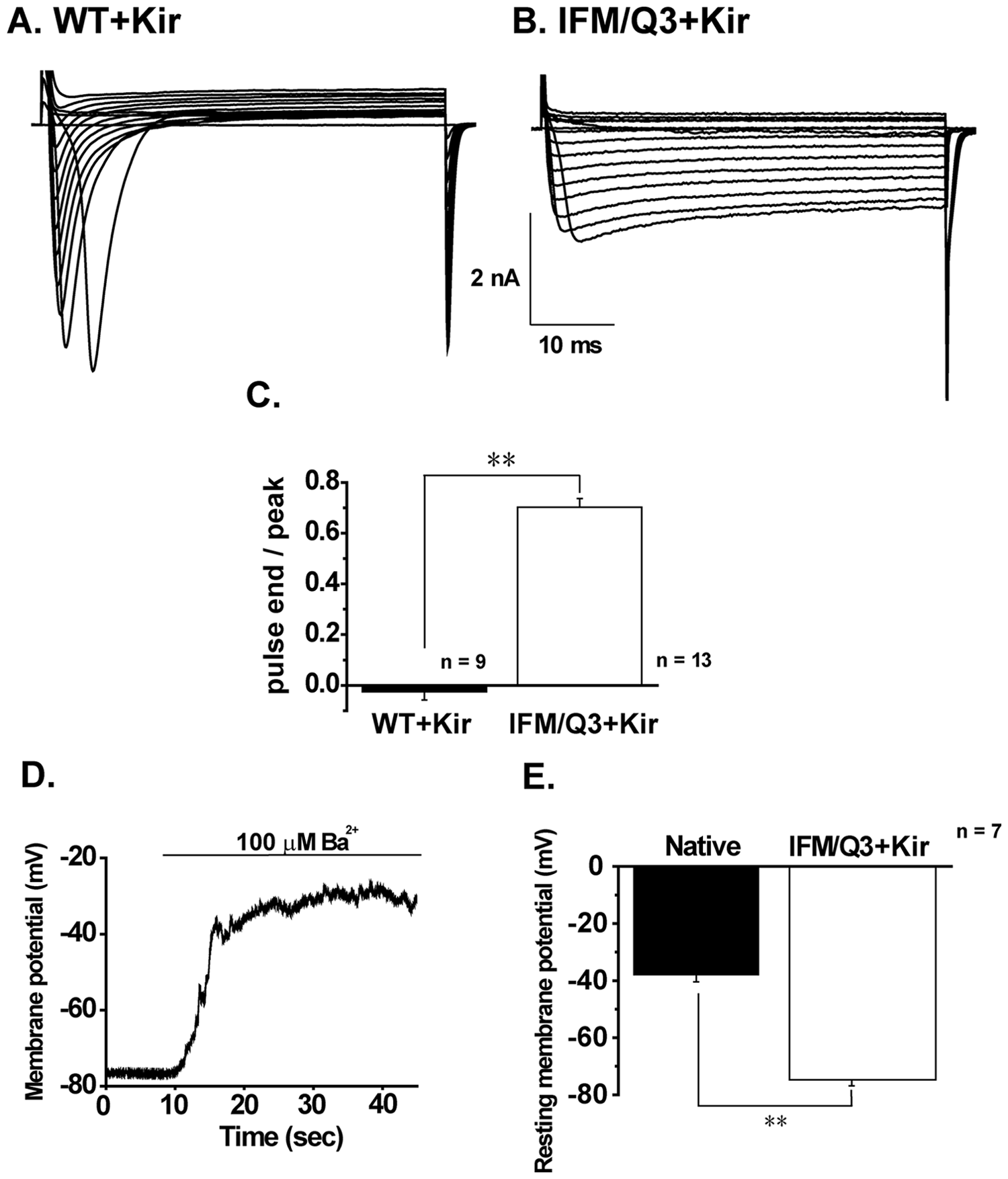
Electrophysiological characterization of HEK293 cells co-expressing Kir2.1 and wild-type or mutated Nav1.5 under whole-cell recording. (
Occurrence of Prolonged AP in IFM/Q3+Kir and Effects of Lidocaine
In the next step, APs elicited by current injection of depolarizing square pulses of 5 ms in duration and 400 pA in amplitude were measured from WT+Kir and IFM/Q3+Kir ( Fig. 2A , B ). APs in WT+Kir showed the peak within 5 ms and progressively turned out within 100 ms ( Fig. 2A ). In contrast, those in IFM/Q3+Kir possessed a long-lasting plateau phase, which gradually declined from +10 to −20 mV and lasted for more than 30 s ( Fig. 2B ). The summarized data of AP duration (APD) indicate significantly prolonged APD in IFM/Q3+Kir (n = 5) compared with that in WT+Kir (n = 5, p < 0.01; Fig. 2C ).
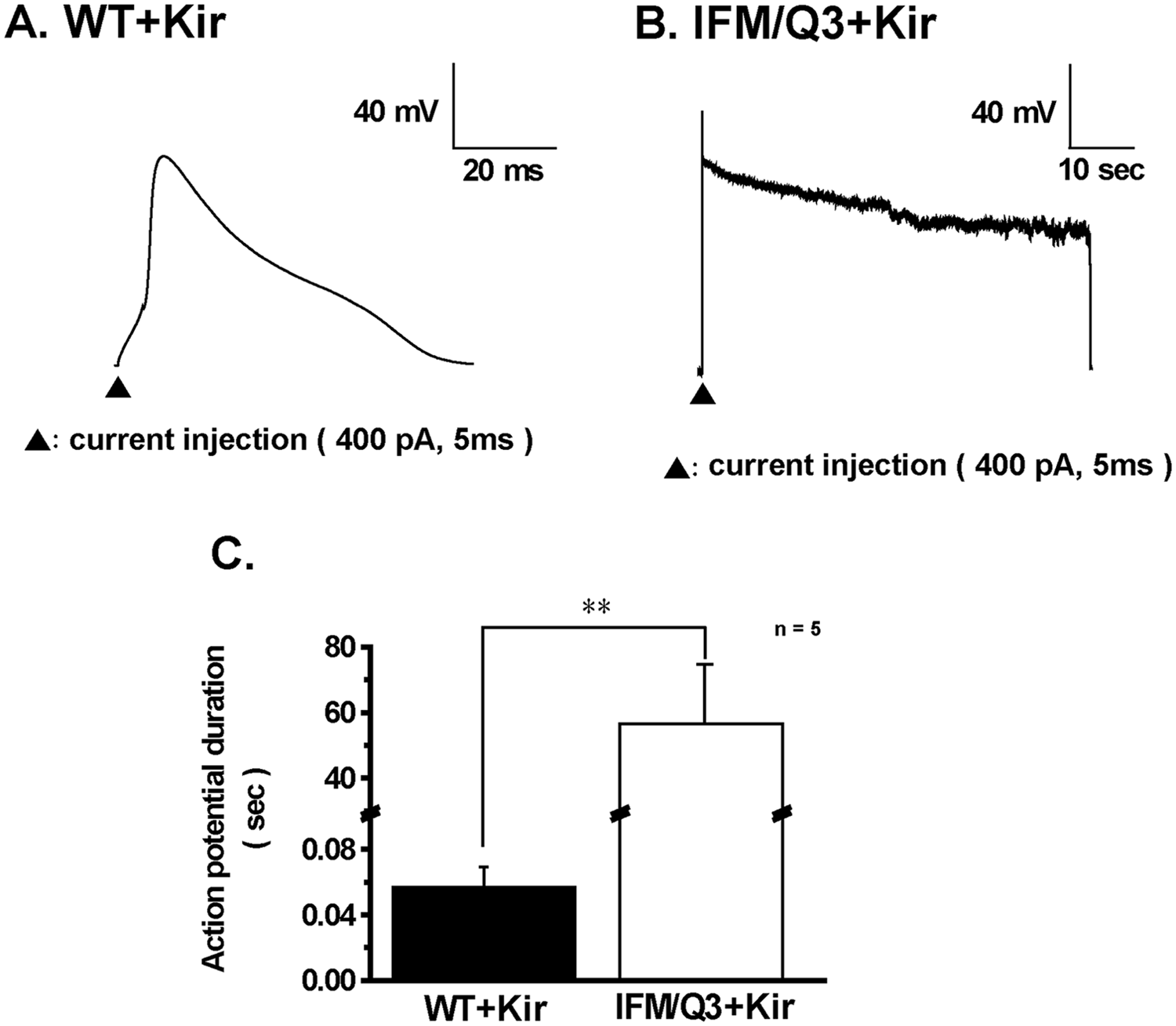
Comparison of action potential (AP) shapes between WT+Kir and IFM/Q3+Kir. (
Effects of lidocaine, a Na+ channel blocker, on APs were examined in WT+Kir and IFM/Q3+Kir ( Fig. 3A, B ). In the presence of 1 mM lidocaine, APs were not elicited by current injection in WT+Kir ( Fig. 3A,b ). Addition of 1 mM lidocaine during a long AP in IFM/Q3+Kir repolarized the cell ( Fig. 3B,a ). Even in the presence of 1 mM lidocaine, electrical responses with shorter duration (<5 s) and smaller amplitude were observed in all IFM/Q3+Kir examined (n = 3; Fig. 3B,b ). Therefore, there appeared a tendency that APs in IFM/Q3+Kir were more resistant to 1 mM lidocaine than those in WT+Kir. To examine the possibility more exactly, the sensitivity of Na+ currents to 1 mM lidocaine was compared between WT+Kir and IFM/Q3+Kir ( Fig. 3C ). Accordingly, the block by lidocaine of Na+ current was significantly smaller in IFM/Q3+Kir (n = 6) than in WT+Kir (n = 6, p < 0.01, respectively).
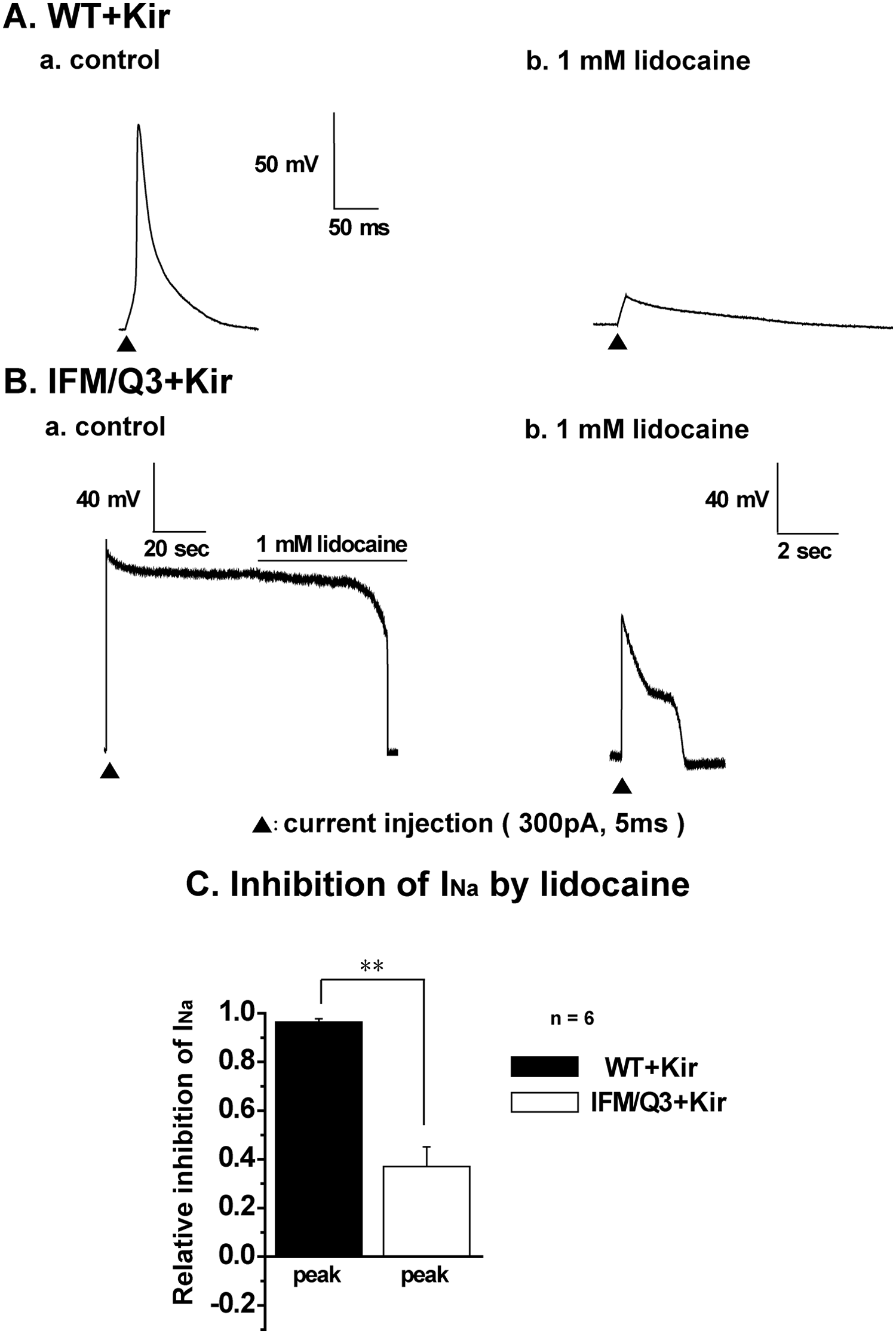
Effects of lidocaine on action potentials (APs) and Na+ current amplitude. (
To measure the prolonged AP in a larger number of cells, membrane potentials were recorded using a slow-response voltage-sensitive fluorescent dye, DiBAC4(3). In IFM/Q3+Kir, a gradual increase in relative fluorescent intensity (F/F 140K ) was observed following the application of a set of three electrical square pulses (arrow, Fig. 4B ) but not in WT+Kir ( Fig. 4A ). The increase in F/F 140K upon ES indicates the occurrence of large depolarization, presumably prolonged AP. The ES-induced increase was not observed in the presence of 1 mM lidocaine, whereas that by 140 mM K+ was detected (not shown). The summarized changes in F/F 140K from a larger number of cells indicate that the increase occurred only in IFM/Q3+Kir but not in WT+Kir ( Fig. 4C , D ).
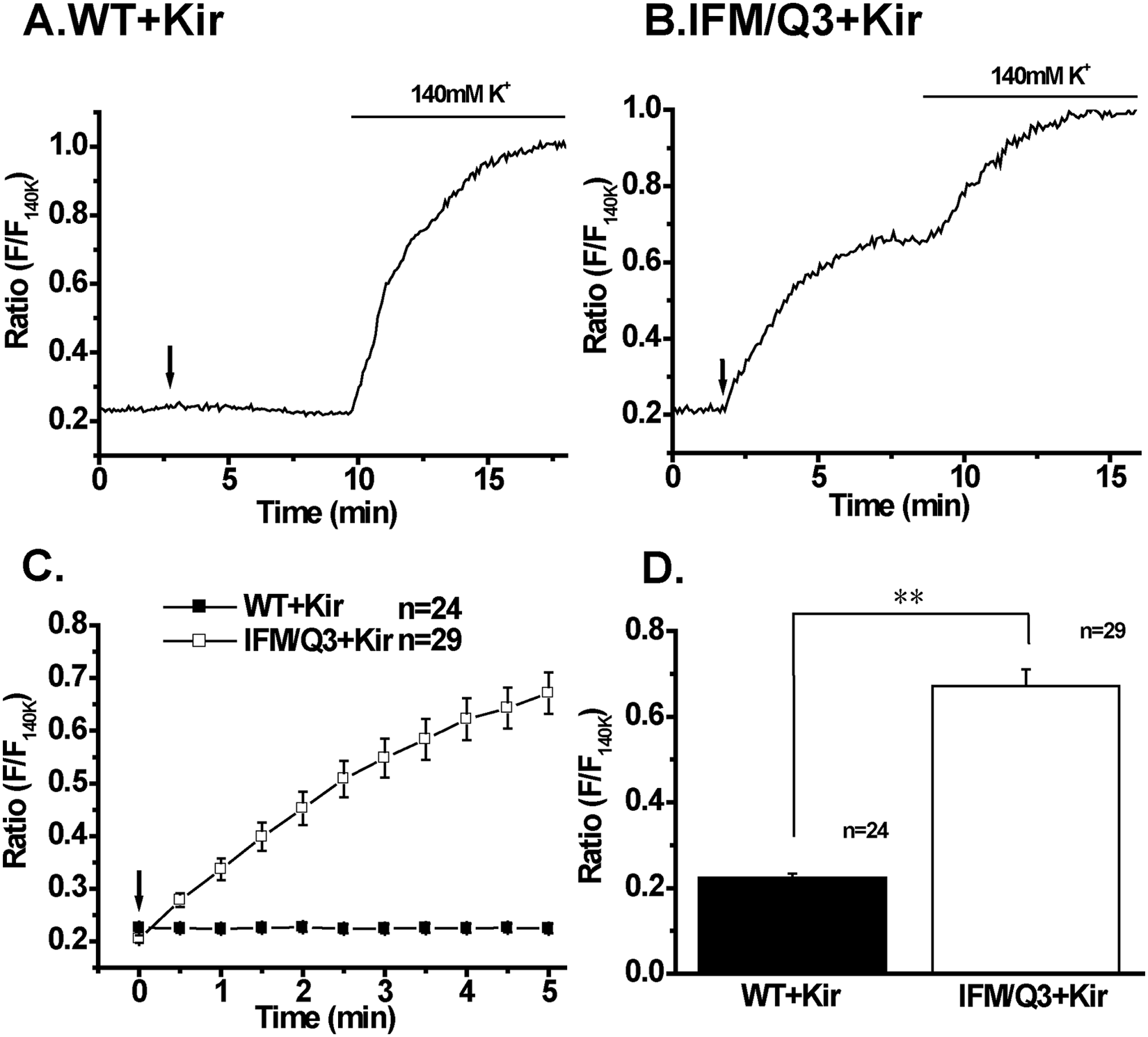
Effects of electrical stimulation (ES) on membrane potential in IFM/Q3+Kir and WT+Kir. Membrane potential changes by ES were monitored as images using DiBAC4(3) and a fluorescent microscope system. (
Detection of Cell Death Induced by a Long AP with MTT Assay
The cell death following application of electrical pulses was examined using 96-well plates and MTT assay. The methods for ES and the process of assay are shown in Figure 5A . Cells cultured in each well were activated by ES, applied three times with an interval of 1 to 3 min, and were again cultured for 2 to 6 h prior to MTT assay. The viability in WT+Kir and IFM/Q3+Kir was approximately 1.0 and 0.15, respectively, when the viability of nonstimulated WT+Kir and IFM/Q3+Kir was taken as 1.0 ( Fig. 5B , C ). The low viability in IFM/Q3+Kir was recovered by the presence of 1 mM lidocaine ( Fig. 5C ). The relative viability of IFM/Q3+Kir in the absence of lidocaine was not apparently affected when the intensity of pulse was ranged from 50 to 200 mA.
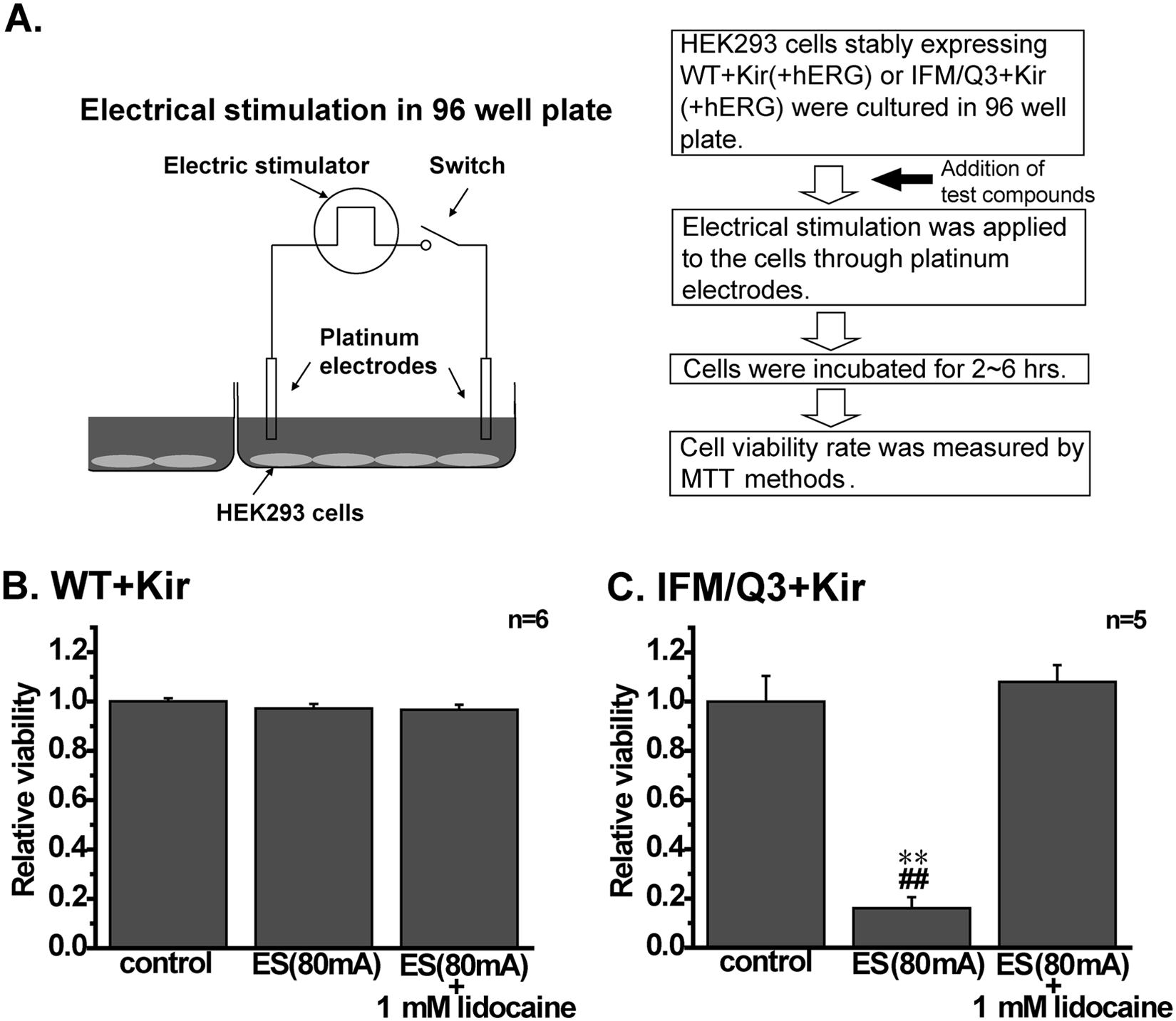
Measurement of cell death induced by electrical stimulation (ES) with MTT assay. (
Co-expression of the hERG Channel for hERG Channel Inhibition Assay
To apply this cell line to a potential drug-safety assay for hERG channel inhibition, we generated novel cells by additionally transfecting IFM/Q3+Kir with hERG (IFM/Q3+Kir+hERG). In these experiments, hERG together with GFP were transiently expressed in IFM/Q3+Kir. If the hERG channels are activated enough to overcome the sustained Na+ current during the plateau phase of AP in IFM/Q3+Kir+hERG, the AP would be turned out, and the cell death would be prevented.
In IFM/Q3+Kir+hERG, application of 10 µM nifekalant markedly reduced the outward current during prepulse at 0 mV and the tail current at −50 mV to a similar extent (see
To measure the prolongation of AP by nifekalant in a larger number of cells, membrane potentials were recorded using DiBAC4(3). Neither the first ES to IFM/Q3+Kir+hERG nor the addition of 10 µM nifekalant per se changed the F/F 140K (left arrow and bar in Fig. 6A). However, the second stimulation in the presence of nifekalant induced a gradual but large increase in F/F 140K . The F/F 140K at 5 min after stimulation was significantly increased by 10 µM nifekalant, suggesting that the block of hERG channel by nifekalant prolonged the AP in IFM/Q3+Kir+hERG (Fig. 6B, C).
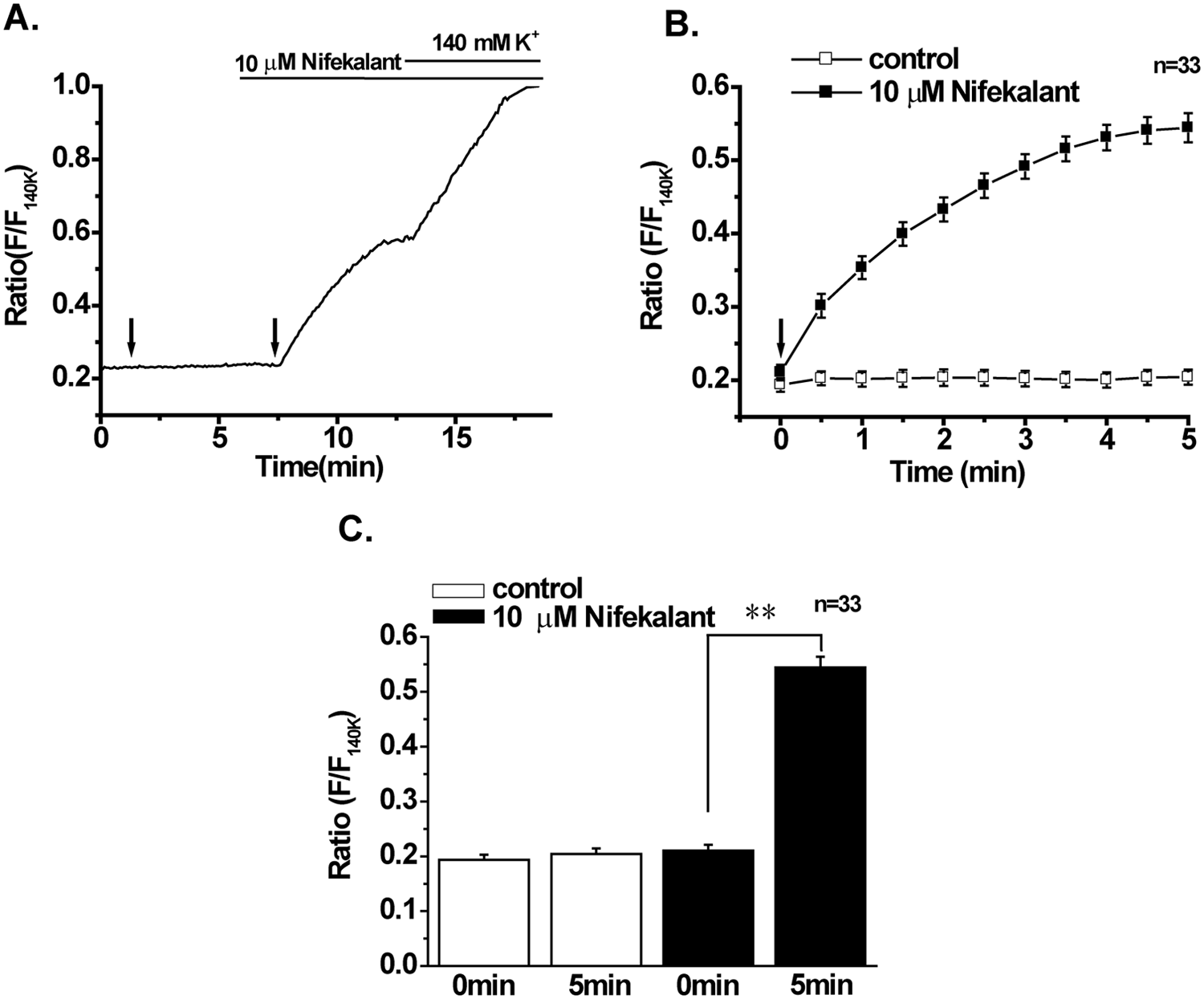
Effects of electrical stimulation (ES) on membrane potential in IFM/Q3+Kir+hERG. Membrane potential changes by ES were monitored using DiBAC4(3) in a similar manner as shown in
Figure 4
. (
In IFM/Q3+Kir+hERG, the cell death induced by ES and effects of 10 µM nifekalant on it were examined. The efficacy of the transient expression of hERG in IFM/Q3+Kir+hERG was high (more than 80%) based on the patch-clamp experiments (not shown). The ES did not significantly affect the viability of IFM/Q3+Kir+hERG (control versus ES in
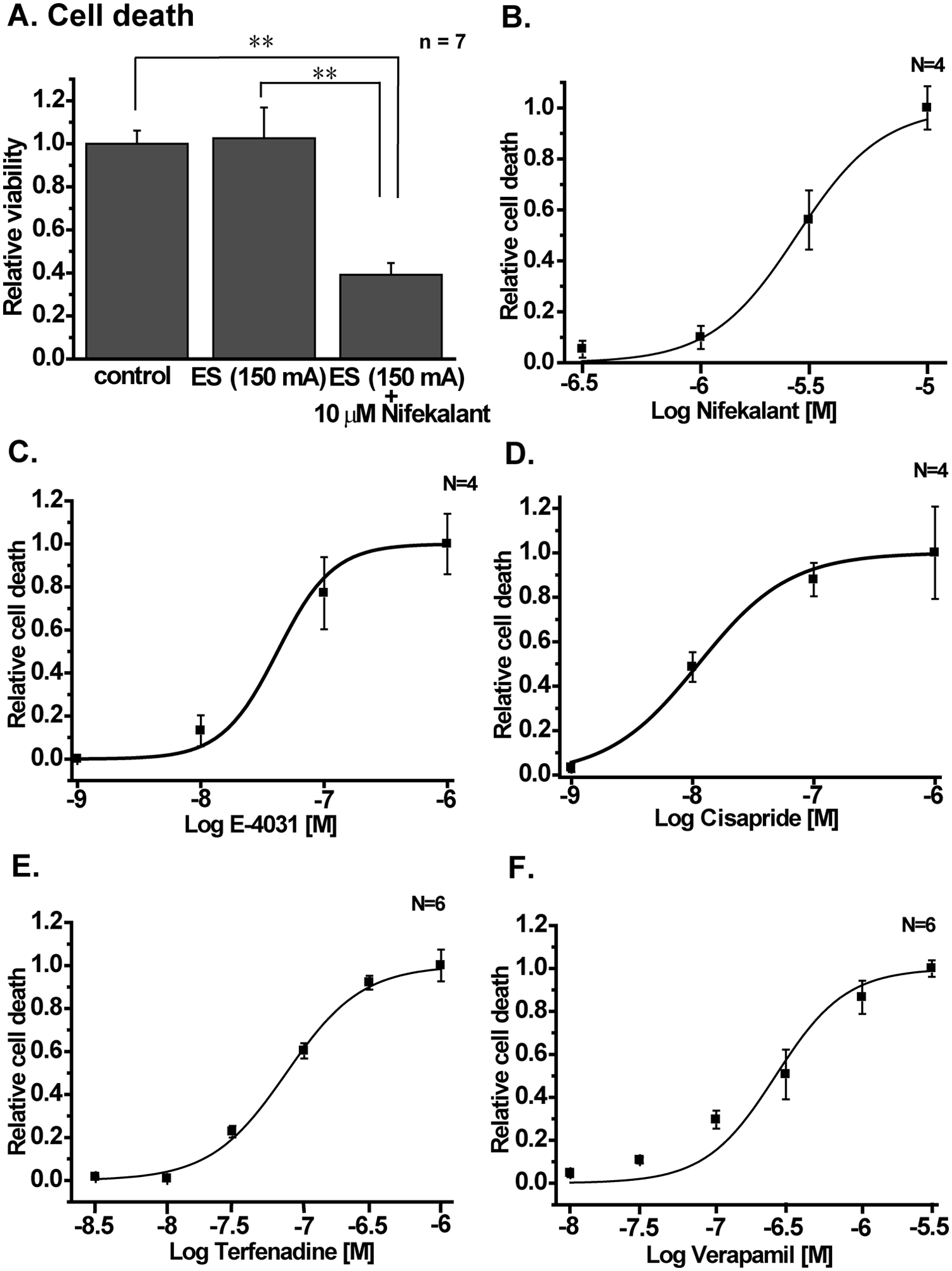
Validating the potency of hERG inhibitors using IFM/Q3+Kir+hERG. (
Effects of four compounds known to block the hERG channel—E-4031, cisapride, terfenadine, and verapamil—were also examined using the system. The EC50 and Hill coefficient were 41.9 ± 16.5 nM and 1.87 ± 0.36 (n = 4) for E-4031 ( Fig. 7C ), 18.9 ± 6.5 nM and 1.16 ± 0.44 (n = 4) for cisapride ( Fig. 7D ), 74.0 ± 2.9 nM and 1.61 ± 0.15 (n = 6) for terfenadine (Fig. 7E), and 275 ± 36 nM and 1.81 ± 0.51 (n = 6) for verapamil (Fig. 7F), respectively. Z′ values for the data were 0.59, 0.51, 0.52, and 0.55, respectively.
The Cell Death by ES Occurred Quickly in IFM/Q3+Kir
To examine the time course of cell death, the capacity to exclude the vital dye trypan blue was measured in IFM/Q3+Kir following ES. The percentages of cells stained by trypan blue were measured by eye in the viewing field of a microscope at 5, 15, 30, and 60 min after the application of ES. The results indicate that the cell death in IFM/Q3+Kir mostly occurred within 5 min from ES (
Discussion
The present results showed that the novel cell line, which was produced by co-expression of Kir2.1 and mutated Nav1.5 (IFM/Q3) in HEK293 cells (IFM/Q3+Kir), caused autoclasis by a prolonged AP occurrence in response to ES. The key issue is that the additional expression of hERG channel to IFM/Q3+Kir markedly shortened APD and significantly prevented cell death induced by ES.
Moreover, this new assay system allows us to quantify the potential of test compounds to inhibit the hERG K+ channel because the hERG block again prolonged evoked APs, resulting in cell death in IFM/Q3+Kir+hERG. The potency of nifekalant, cisapride, E-4031, terfenadine, and verapamil, which have been known as hERG channel blockers, to induce cell death was evaluated in IFM/Q3+Kir+hERG. The EC50s of these drugs were 2.7 µM, 19 nM, 42 nM, 74 nM, and 275 nM, respectively, and these values are close to their EC50s for hERG K+ current in heterologous expression system; 7.8 µM, 28 nM, 26 nM, 45 nM, and 143 nM, respectively.16–18 In contrast, 10 µM nifedipine did not affect the cell death (
Co-expression of Kir2.1 with IFM/Q3 is essential for this assay system to keep the resting membrane potential negative to −75 mV, because the noninactivating Na+ current through IFM/Q3 is large enough to kill HEK293 cells at less negative resting membrane potentials.14,15 Actually, 100 µM Ba2+, a blocker of Kir2.1, induced marked membrane depolarization in IFM/Q3+Kir (
Fig. 1D
) and killed the cells without ES (EC50 of 14.5 ± 2.3 µM, n = 6;
Moreover, a test compound, which blocks both Nav1.5 and hERG, will prevent cell death of IFM/Q3+Kir+hERG upon ES and could be evaluated as false-negative. Quinidine blocks both hERG and Nav1.5 in cardiac myocytes, but the effect on hERG appears at lower concentrations.
19
Accordingly, cell death of IFM/Q3+Kir+hERG upon ES was observed in the presence of quinidine at low concentrations (<3 µM) and the inhibition of cell death at higher concentrations (
The cause of cell death in IFM/Q3+Kir upon ES is not clear in this study but is presumably the rapid increase in intracellular Na+ concentration ([Na+]i), because the block of sustained Na+ influx by lidocaine prevented cell death. In previous studies, the persistent opening of a sodium channel by treatment with veratridine causes the elevation of [Na+]i, which perturbs ion homeostasis and triggers cell death in the HEK cells expressing Na+ channel and the neuronal cells.20,21 The staining of IFM/Q3+Kir by trypan blue revealed that more than 90% of cell death occurred within 30 min after the application of ES (
Long QT syndrome upon medication occasionally leads to a delay in cardiac AP repolarization and predisposes patients to potentially fatal arrhythmias.1,3 Consequently, evaluating the effect of new drugs on hERG function is now a mandatory step in the development of novel pharmaceuticals.
22
The assessment of compound activities on hERG channels is conducted with use of an automated, electrophysiological, ion flux method and so on.7,23 The advantages of the hERG channel safety assay by use of IFM/Q3+Kir+hERG can be listed as follows: (1) The procedure and results are so simple that the assay including analyses can be performed without electrophysiological techniques. (2) The initial cost for equipment and running cost are low. In this study, cell death was evaluated by the most popular MTT methods. (3) This assay fits well to HTS because of the low cost and ease of automation. The total time required for evaluation mainly depends on the incubation time after ES and the period during which cells are dying, maybe <30 min (
There are several options for the use of IFM/Q3+Kir and IFM/Q3+Kir+hERG in assays. Instead of ES, application of 25 to 45 mM K+ solution induced cell death in IFM/Q3+Kir (not shown). In HEK293 cells co-expressing Kir2.1 and T-type Ca2+ channel, Ca2+ channel was activated by high K+. 25 The hERG inhibition assay by use of DiBAC4(3) as HTS has been applied using CHO cells expressing hERG and high K+ solution for hERG channel activation. 6 The advantage of this new method is as follows. (1) Test compounds are evaluated by their effects on AP occurrence triggered by ES in IFM/Q3+Kir+hERG, suggesting that the state- and/or voltage-dependent effects can be detected. (2) The viability of IFM/Q3+Kir+hERG upon ES is determined by AP occurrence in an all-or-none manner within 30 min after ES. Therefore, this system easily fits to HTS by developing a 96-/384-well plate, in which ES can be applied at one time.
In addition to the application of IFM/Q3+Kir to the hERG K+ channel inhibition assay, it may be available for evaluation of new compounds acting on other voltage-dependent K+ channels, such as Kv1.3, Kv1.5, or KCNQ1+KCNE2. It may even be available for the screening of compounds acting on ligand-gated ion channels. The advantage of this system against other simple cell systems such as yeast 26 may be that the validation using AP allows us to evaluate state-dependent effects on voltage-sensitive ion channels.
In conclusion, the new assay system for hERG channel inhibition using IFM/Q3+Kir+hERG is a novel tool because of its simplicity, accuracy, and high cost performance. Further application of methods for the screening of drug-development targeting on ion channels will be also expected. The technology developed in this study has been applied to patent filings (PCT/JP2011/064967), and the laid-open disclosure number is WO2012/002460.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This investigation was supported by a Grant-in Aid for Scientific Research (B) (20390027 and 23390020) and a Grant-in-Aid for Exploratory Research (23659046) from the Ministry of Education, Culture, Sports, Science, and Technology to Y.I.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.