
Abstract
Friedreich ataxia (FRDA) is an autosomal recessive neuro- and cardiodegenerative disorder for which there are no proven effective treatments. FRDA is caused by decreased expression and/or function of the protein frataxin. Frataxin chaperones iron in the mitochondrial matrix for the assembly of iron–sulfur clusters (ISCs), which are prosthetic groups critical for the function of the Krebs cycle and the mitochondrial electron transport chain (ETC). Decreased expression of frataxin or the yeast frataxin orthologue, Yfh1p, is associated with decreased ISC assembly, mitochondrial iron accumulation, and increased oxidative stress, all of which contribute to mitochondrial dysfunction. Using yeast depleted of Yfh1p, a high-throughput screening (HTS) assay was developed in which mitochondrial function was monitored by reduction of the tetrazolium dye WST-1 in a growth medium with a respiration-only carbon source. Of 101 200 compounds screened, 302 were identified that effectively rescue mitochondrial function. To confirm activities in mammalian cells and begin understanding mechanisms of action, secondary screening assays were developed using murine C2C12 cells and yeast mutants lacking specific complexes of the ETC, respectively. The compounds identified in this study have potential relevance for other neurodegenerative disorders associated with mitochondrial dysfunction, such as Parkinson disease.
Introduction
Friedreich ataxia (FRDA) is an autosomal recessive neuro- and cardiodegenerative disorder, with a prevalence of approximately 1 in 40 000 in European populations (see recent review by Dr. Massimo Pandolfo 1 ). FRDA is characterized by progressive ataxia of all four limbs, dysarthria, areflexia, sensory loss, and muscle fatiguability. Skeletal deformities and cardiomyopathy are found in most patients, impaired glucose tolerance and diabetes mellitus are found in ~30% of patients, and reduced visual acuity and hearing loss are occasionally seen. 2 Onset of symptoms usually occurs around puberty, and most patients are confined to a wheelchair by their late 20s. Myocardial failure is the most common cause of premature death.
FRDA is caused by mutations in the nuclear gene, FXN, which encodes the highly conserved protein frataxin. 3 Frataxin localizes primarily to the mitochondrial matrix, where it chaperones iron for iron–sulfur cluster (ISC) assembly.4,5 ISCs are prosthetic groups important for the function of many proteins, both mitochondrial and cytosolic, including aconitase and mitochondrial respiratory complexes I, II, and III. Decreased frataxin function in yeast models, mouse models, and humans with FRDA is associated with mitochondrial iron accumulation, mitochondrial dysfunction, and increased oxidative stress.6-18 Because iron dysregulation and mitochondrial dysfunction underlie the signs and symptoms of FRDA, identification of small molecules that improve mitochondrial function in cell-based models could lead to effective drugs for FRDA and may have implications for other neurodegenerative diseases in which iron dysregulation and/or mitochondrial dysfunction play a role.
Yeast are permeable to compounds in chemical libraries and have been used in numerous screens. Virtually every finding in the yeast model system of FRDA has been confirmed in conditional knockout mice, primary human fibroblasts, and patients with FRDA. Frataxin and Yfh1p, the yeast frataxin homologue, are functional homologues, and the connection between FRDA and iron metabolism was discovered in yeast. The strikingly close correspondence between the biochemical abnormalities in yeast with mutations in Yfh1p, as well as mammalian cells with mutations in the gene encoding frataxin, further supports the suitability of the yeast model system for high-throughput compound screening. Mitochondrial function can be assayed in yeast by quantifying growth in media containing a nonfermentable carbon source, such as glycerol. A colorimetric assay using a tetrazolium dye such as WST-1, which is reduced by cellular dehydrogenases, can then be used to quantitatively monitor the metabolic activity of the cells.
The aim of the high-throughput screening (HTS) assay described herein was to identify compounds that can improve mitochondrial function in yeast lacking Yfh1p grown in a nonfermentable growth medium. The yeast Yfh1p expression construct used in this study has a galactose-inducible, dextrose-repressible promoter driving Yfh1p expression. 4 Yfh1p is expressed when the yeast are grown in the presence of galactose and inhibited when they are grown in the presence of dextrose. Growing the cells in the presence of dextrose turns off the expression of Yfh1p and leads to the depletion of cellular Yfh1p over time. Transferring the Yfh1p-depleted cells into a nonfermentable, glycerol-based medium compels mitochondrial respiration. Adding drugs that can bypass the lack of Yfh1p leads to recovered mitochondrial respiratory function as demonstrated by the metabolism of the tetrazolium dye, WST-1. WST-1 is a stable reagent, often used to monitor cell proliferation, and exhibits a good linear range and accelerated color development. Cleavage of the tetrazolium salt to formazan is accompanied by an increase in absorbance at 450 nm.
A secondary screening assay in murine C2C12 cells was developed using iron overload and inhibition of antioxidant response to recapitulate the FRDA phenotype. The aim of this assay was to confirm activities in mammalian cells. Another secondary screening assay was developed using yeast mutants lacking specific complexes of the electron transport chain (ETC). The aim of this assay was to begin to understand mechanisms of action, specifically to determine whether hit compounds that increase adenosine triphosphate (ATP) in cells depleted for Yfh1p act through the ETC.
Materials and Methods
Libraries and Compounds
The Yfh1p yeast assay was screened against three libraries: 1120 biologically active compounds (1 mM; Microsource Discovery Systems, Inc. [Gaylordsville, CT] Spectrum Collection), the 100 000–compound National Institute of Neurological Disorders and Stroke (NINDS) library (5 mg/mL; comprising the ~50 000–compound ChemBridge [San Diego, CA] CNSSet library and another ~50 000–compound diversity set from Tripos [St. Louis, MO] of primarily derivatized heterocycles), and a third 5000 diversity subset (1 mg/mL) generated from the NINDS 100 000–compound library. Compounds in the NINDS diversity subset were selected to serve as a representative sample of the overall chemical diversity of the large library. All compounds were solubilized in 100% DMSO and diluted to working concentration in media immediately before addition to assay plates. The final concentration of DMSO was maintained between 0.2% and 1% to achieve a final test concentration for compounds and positive control of either 10 µM or 10 µg/mL. The compounds used in the secondary screening assays (i.e., in murine C2C12 cells and in yeast mutants lacking specific complexes of the ETC) were obtained from ChemBridge.
Cell Lines, Expansion of Culture, and Yeast Strains
The yeast strain used for the primary HTS assay was generously provided by Dr. Roland Lill and has been described. 4 Briefly, the galactose-inducible, dextrose-repressible promoter, GAL1-10, was used to replace the endogenous Yfh1 promoter using PCR-mediated gene replacement. Integration at the correct chromosomal locus was verified by PCR, and the inducibility/repressibility was confirmed by Western analysis using antibodies to Yfh1p. For the assay, a stock culture was streaked on a galactose agar plate and incubated for 3 days at 30 °C. A single colony was inoculated in 10 mL induction medium (“YpGal”) for 24 h using galactose as a carbon source (Yfh1 on). Cells were transferred to repression medium using dextrose as a carbon source (CSMM-D; Yfh1 off) and incubated for 24 h to allow for Yfh1p protein decay. Cells were washed 2× in CSMM-D and resuspended to 2 × 105 cells/ml in CSMM-D. Following growth in CSMM-D, cells were washed 2× in CSMM-Gly and then resuspended at 2 × 108 cells/mL in CSMM-Gly containing 15% glycerol and rate-frozen to −80 °C. Aliquots of stock culture were stored at −80 °C prior to use. The stock culture was tested for bacterial contamination on sheep blood agar (SBA) and glycerol agar plates, yielding negative results. Before use in a screen, frozen stocks were also evaluated for assay performance by coefficient of variation (CV) and response to control compounds. For screening, cells were washed 1× in CSMM-Gly media and resuspended to 2 × 106 cells/mL in CSMM-Gly and grown overnight with shaking at 30 °C. At this point, Yfh1p expression is off, and the glycerol carbon source restricts the cells to respiratory metabolism. After 24 h, in CSMM-Gly media, the cells were pelleted and resuspended to 4 × 106 cells/mL in the same media for the assay.
Yeast strains lacking Atp15p, which is necessary for the function of complex V in the ETC, and Ndi1p, the yeast equivalent of complex I in the ETC, were obtained from the Saccharomyces cerevisiae haploid knockout strain collection (Invitrogen, Carlsbad, CA). The Yfh1p haploid knockout strain FY 1679-05A (mating type
The murine myoblast cell line, C2C12, was cultured in standard medium (Dulbecco’s modified Eagle’s medium [DMEM] supplemented with 10% fetal bovine serum [FBS] and 1% penicillin/streptomycin). In total, 4000 cells/well were seeded into 96-well, white opaque tissue culture plates. Ferric ammonium citrate (FAC; F-5879 [Sigma, St. Louis, MO], iron content 16.5%−18.5%) was added 4 h later to a final concentration of 100 µg/mL. The following day, L-buthionine (S,R)–sulfoximine (BSO) was added to 100 µM final concentration, and 2 h later, compounds were added. The final concentration of DMSO in the assay was 1%. Twenty-four hours later, the cells were washed with phosphate-buffered saline (PBS) and treated with the CellTiter-Glo assay kit (Promega, Madison, WI) as per the manufacturer’s instructions. Idebenone was from Sigma (compound I5659) and tannic acid was from Fisher Scientific (compound S80215; Fisher Scientific, Waltham, MA).
For the ATP “burst” assay, yeast knockout strains were grown in YPD medium and diluted as appropriate to log-phase growth at the time of assay. Cells were spun down, washed in water, and resuspended in SC-Gly medium. Compounds resuspended in DMSO were used at a 100-µM final concentration. The final DMSO concentration was 1%. The cells were lysed by addition of an equal volume of BacTiter-Glo assay reagent (Promega).
Validation of the Primary Screening Assay
Validation of the primary HTS assay included optimizing cell densities, well volume, incubation times, humidified versus nonhumidified chambers, and determination of the highest concentration of DMSO that would not affect cell viability. These conditions were determined to be 52 000 cells per well in a total volume of 25 µL containing 10% WST-1 and 10 µM or 10 µg/mL compound. Incubation was for 4 days in humidified containers. DMSO could be tolerated up to 4% without adversely affecting the assay. To identify a commercially available positive control compound, the Microsource library, consisting of 1120 biologically active compounds, was screened using yeast lacking Yfh1p. Several active compounds were identified, and EC50s and color interference with WST-1 were evaluated. Tannic acid was selected to serve as the positive control. Once these conditions were optimized and a control was identified, the assay was further validated through variability testing. Z′ analyses were done to evaluate the assay’s dynamic range and the coefficient of variation associated with the signal measurements. To determine if the assay could identify hits with confidence, the 5000-compound diversity set was then screened in duplicate to evaluate reproducibility. During this pilot screen, a number of false positives were observed. Sporadic low-level bacterial contamination was identified as the source. Penicillin and streptomycin were evaluated for their effect on the assay and their ability to minimize the false positives. Results showed that the antibiotics did not affect the Yfh1p assay but did reduce the false-positive rate. For the screen, Gibco (Carlsbad, CA) penicillin/streptomycin for tissue culture was used at 1% in the assay media.
HTS Yfh1p Yeast Assay
Compounds were added via a dilution step in 12 µL assay medium at 2× the final concentration to black, clear-bottom, tissue culture–treated 384-well plates using the Biomek FX liquid handling system (Beckman Coulter Instruments, Brea, CA). Immediately following compound addition, 2.5 µL (1:10) of Roche WST-1 Cell Proliferation Reagent (Roche, Basel, Switzerland) was added using the Biomek FX. Yfh1p yeast were then added at a density of 4 × 106 cells/mL in 13 µL assay media, using the Matrix (Maumee, OH) WellMate, and incubated in a humidified chamber at 30 °C for 4 days. After a 96-h incubation, absorbance was measured at wavelengths 450 nm and 630 nm (to estimate growth) with a PerkinElmer (Waltham, MA) EnVision plate reader. To screen 100 000 compounds efficiently, a batch of one hundred 384-well plates was processed daily for 4 consecutive days. Several quality control (QC) steps were implemented to ensure the consistency between daily screening runs. A daily QC plate was run with each batch in addition to using a laboratory information management system (LIMS) to monitor lot numbers of the reagents used. Daily QC checks to confirm accurate volume dispensing for cell plating and compound and reagent addition were also performed.
Data Analysis
During validation, a number of data analysis methods were evaluated. Initially, the WST-1 signal (OD450) and turbidity (OD630) were both read. To compensate for plating differences and cell growth, data were normalized, dividing OD450 by OD630. We also used % Recovery as the output, calculated as % Recovery = [(normalized test well value – average normalized cell control)/(average normalized cells with control drug – average normalized cell control)] × 100. During development, very little variation was observed in the OD630 read, so this normalization was discontinued (i.e., under the conditions of this assay, cell proliferation was negligible, and hence any increase in WST-1 was not due to an increase in cell number). In the transition from validation to the screening campaign, there was a notable increase in the variability of the tannic acid control. Calculating % Recovery based on the tannic acid signal became problematic and was also discontinued. Because the cell signal was very consistent, we adopted fold induction for analysis of compound data. At the end of each screen, data were imported and processed using IDBS (Guildford, UK) Activity Base. Fold induction was calculated by dividing the WST-1 reduction for a test compound by the WST-1 reduction of the median of the cell control (which comprised cells plus DMSO as carrier control).
Results
Validation
A compound library consisting of 1120 biologically active compounds was screened against yeast Yfh1p ( Fig. 1 ). Using a cutoff of 3 SD above the median, we identified 10 hits: menadione, tannic acid, sennoside B, epigallocatechin 3,5-digallate, theaflavin monogallate, theaflavin digallate, katacine, 2,2-bisepigallocatechin monogallate, pararosaniline pamoate, and pyrvinium pamoate. Tannic acid is found in tea and red wine, and epigallocatechin 3,5-digallate, theaflavin monogallate, theaflavin digallate, and 2,2-bisepigallocatechin monogallate are all flavanols, which are redox-active free-radical scavengers found in green tea, red wine, berries, and chocolate.19,20 Catechins, gallates, and other flavanols are also known to chelate iron.21-23 Menadione (vitamin K3) is a synthetic, water-soluble vitamin K with a parabenzoquinone structure similar to that of CoQ10 and idebenone, which are lipid-soluble antioxidants under evaluation in clinical trials for FRDA.14,24-24
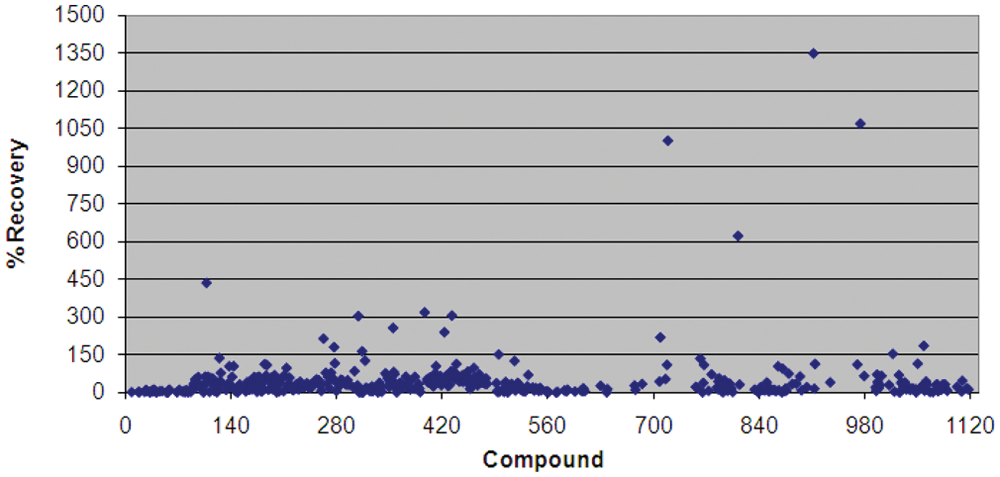
Validation screen of the Microsource Spectrum collection of 1120 biologically active compounds screened at 10 µM: scatter plot of % Recovery from the metabolic block created by decreased expression of Yfh1p. % Recovery is calculated as [(normalized test well value – average normalized cell control)/(average normalized cells with control drug – average normalized cell control)] × 100. Raw absorbance data, from increased WST-1 reduction, were normalized to cell density by dividing the OD450 by OD630. Cell control is equivalent to 0% recovery, and a known active compound, octyl gallate, was used to calculate a 100% recovery level. Several compounds in the validation screen produced a more robust response than the control compound, which is reflected in % Recoveries greater than 100%.
A dose–response assay was performed for four of the active compounds identified. Three of the compounds exhibited a clear dose response, whereas the fourth showed toxicity at higher concentrations. Tannic acid showed the most consistent and reproducible results and was chosen as the positive control for this assay (
Fig. 2
). Several Z′ and CV plates were run with tannic acid and/or DMSO, giving consistent and acceptable results. A color control experiment was performed to demonstrate that the compounds did not interfere colorimetrically with the WST-1 end point. The 5000-compound (5K) diversity set was then screened in duplicate. In the first 5K run, Z′ values ranged from 0.73 to 0.9. In the second 5K run, Z′ values ranged from 0.56 to 0.8. The results from the two replicates showed very good reproducibility based on the Spearman correlation (
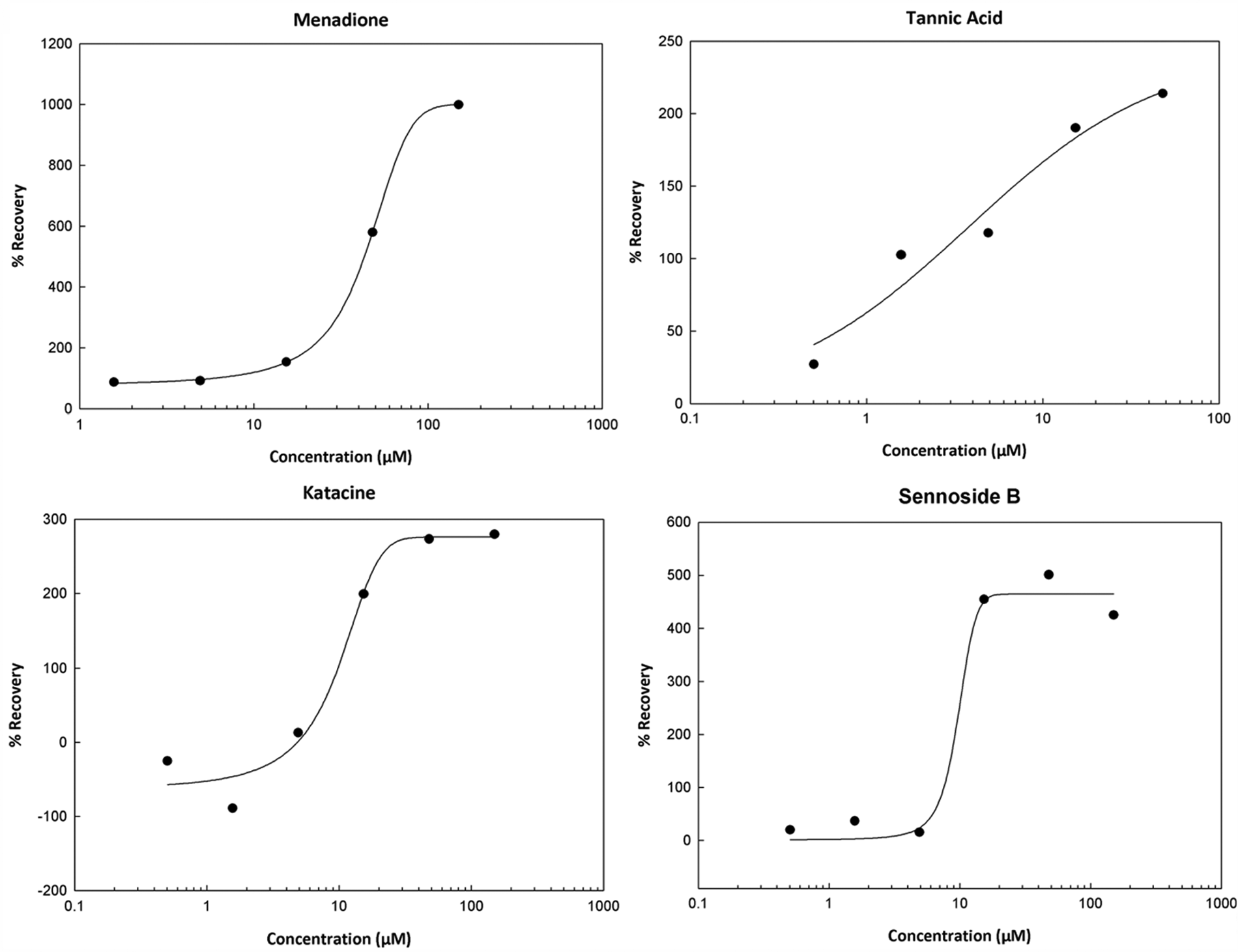
Validation screen of the Microsource Spectrum collection of 1120 biologically active compounds screened at 10 µM: dose–response graphs for four of the active compounds identified in the screen. Each compound was screened in triplicate, and % Recovery was calculated, as described in the text and in the Figure 1 legend. EC50 values were calculated using ExcelFit. EC50 values and Hill slopes were as follows: menadione 62.85 µM, 1.24; tannic acid 3.81 µM, 0.79; sennoside B 8.26 µM, 6.45; and katacine 9.17 µM, 2.35.
HTS Yfh1p Yeast Assay
A new yeast stock was grown, aliquoted, and frozen for the full 100 000–compound (100K) library screen. This stock culture was tested for contamination on SBA and glycerol agar plates, yielding negative results. Z′ and CV plates were run on the new stock, yielding acceptable results (Z′ = 0.6, CV = 4%–14%). However, the 100K run results showed greater variability with tannic acid than we had seen during the validation phase (
Secondary Screening
We tested a number of cell lines to develop a secondary mammalian-cell screening assay. Our most successful system uses the murine myoblast cell line C2C12. To recapitulate the FRDA phenotype, we first tested the effects of overloading the C2C12 cells with iron, using FAC; inhibiting the rate-limiting step of glutathione synthesis, using BSO; and combining the two at sublethal concentrations for each. Approximately 4000 cells per well were plated in standard medium in 96-well plates, and FAC was added 4 h later. The cells were incubated overnight (20 h) in standard tissue culture incubators, after which BSO was added. After another overnight incubation (24 h), ATP content was determined using the CellTiter-Glo assay (Promega).
Our goal was to find conditions that leave 10% to 20% of the cells viable. (ATP content was essentially a surrogate measure of viability, as was evident by microscopic examination of the cells.) The rationale for 10% to 20% viability was to leave a sufficient difference from control conditions to discern a wide range of drug effects but without making the conditions too harsh to discern any drug effects at all. The combination of 100 µg/mL FAC and 100 µM BSO decreased ATP content just over sevenfold (
To validate our secondary C2C12 cell screening assay, we tested our HTS control compound, tannic acid, as well as the parabenzoquinone idebenone, which is under evaluation in clinical trials for FRDA.24-27 Both compounds rescued C2C12 cells from loss of viability due to treatment with FAC and BSO ( Fig. 3 ). Idebenone was efficacious at concentrations of 100 nM and 1 µM; tannic acid was efficacious at 100 µM but apparently toxic at 1 mM ( Fig. 3 ). (Dose–response analyses of tannic acid and assessment of its toxicity, both in yeast and in C2C12 cells, were problematic due to precipitation at higher concentrations and/or interference with assay readouts at higher concentrations secondary to its dark color in solution.)
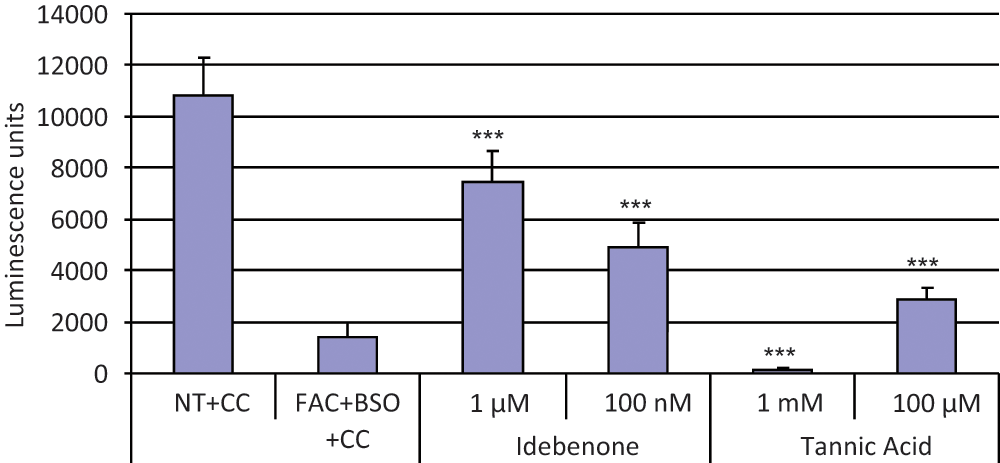
Secondary screening in mammalian cells: idebenone and tannic acid rescue C2C12 cells treated with ferric ammonium citrate (FAC) and L-buthionine (S,R)–sulfoximine (BSO). C2C12 murine myoblast cells were seeded in 96-well plates and treated with a combination of 100 µg/ml FAC and 100 µM BSO (columns 2–6) for 48 h and 24 h, respectively, as described above. Two hours after the addition of BSO, idebenone at a final concentration of 1 µM or 100 nM (columns 3 and 4, respectively) or tannic acid at a final concentration 1 mM or 100 µM (columns 5 and 6, respectively) was added to the cells. Drug carrier (DMSO) final concentration was 1%. After 24 h, cells were lysed and their viability assessed by chemiluminescence measurement of intracellular adenosine triphosphate (ATP; see Methods). ATP content is shown on the ordinate in (arbitrary) luminescence units. Idebenone at both concentrations rescued the C2C12 cells treated with FAC and BSO (columns 3 and 4). Tannic acid at 100 µM rescued the C2C12 cells treated with FAC and BSO; however, tannic acid decreased the viability of the cells at 1 mM (columns 5 and 6, respectively). The data shown are the mean and the standard deviation calculated on eight replicates. The experiment shown is representative of at least two independent experiments. CC, carrier control (DMSO at a final concentration of 1%); NT, no treatment. ***Two-sided p < 0.001 compared with carrier control (using Student t test).
A structural analysis of the 122 highest potency hit compounds (out of the 302 identified) revealed 37 compounds with 9 common core structures ( Fig. 4 ). Two of the core-structure groups ( Fig. 4 ; groups 4 and 5) are similar to the parabenzoquinones idebenone and CoQ10. Core-structure group 9 ( Fig. 4 ) contains a moiety similar to that seen in flavinoids, such as the four flavins and catechins identified in our initial screen of 1120 biologically active compounds (epigallocatechin 3,5-digallate, theaflavin monogallate, theaflavin digallate, and 2,2-bisepigallocatechin monogallate). None of the 9 common core structures is particularly similar to tannic acid, a polyphenol with a molecular weight of > 1700. Core structures 1 through 3 and 6 through 8 are, to our knowledge, novel.
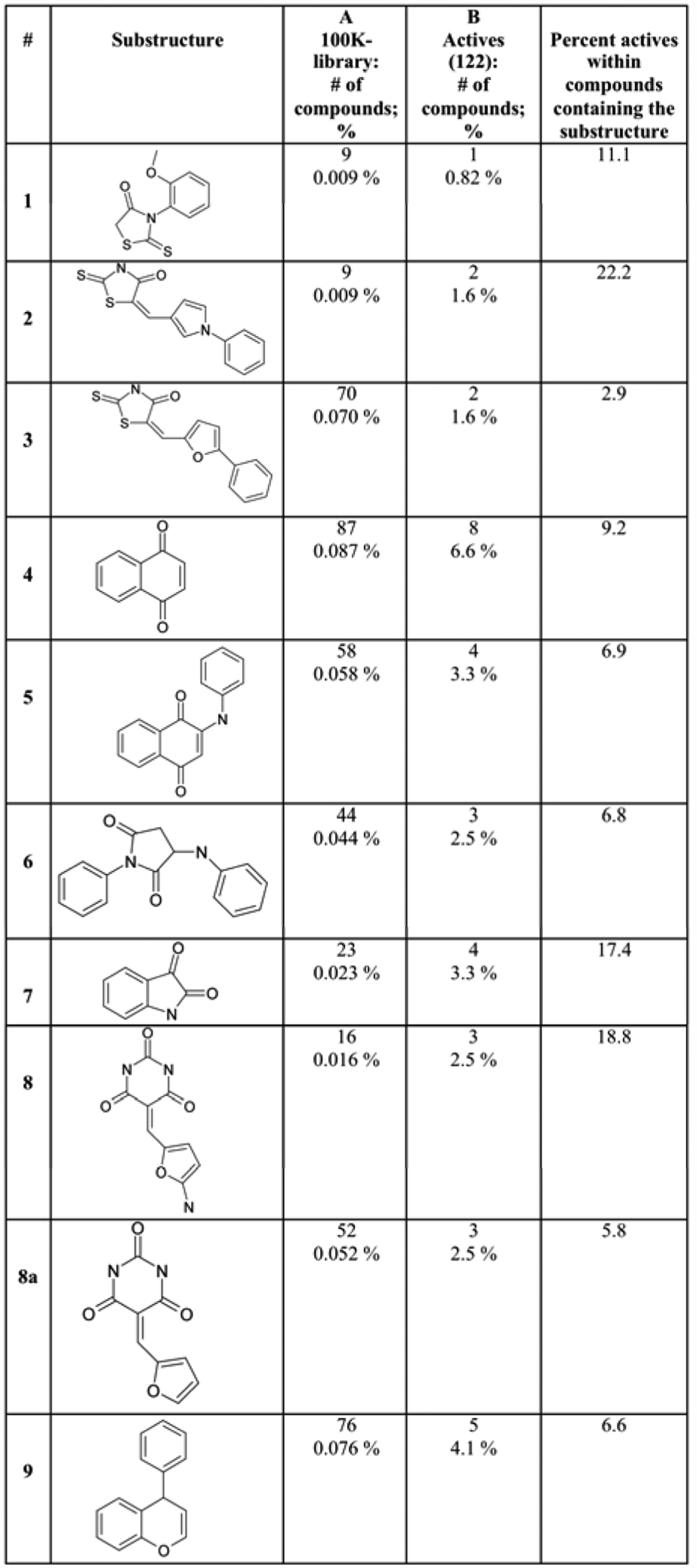
Common core structures from among the 122 highest potency hit compounds (out of the 302 identified), with EC50s <50 µg/mL. Columns A and B list the number and percentage of compounds containing each scaffold within the 100K library and the 122 highest potency hit compounds, respectively.
Several representative compounds (
Fig. 5
) from these core-structure groups are active in our mammalian-cell model, as indicated by increased ATP content and improved viability (
Fig. 6
and data not shown). Representative secondary screening data for one of the parabenzoquinone core-structure groups, group 5, is shown in
Figure 7
. (Compound 5-1 was identified in the primary screen; analogues of 5-1—compounds 5-2, 5-3, and 5-4—were obtained from ChemBridge.) Both compound 5-2 and compound 5-4 increased ATP more than compound 5-1 (p = 0.002 and p = 0.01 by two-sided Student t test, respectively). Compound 5-2 increased ATP more than compound 5-3 (p = 0.008 by two-sided Student t test) despite only a very subtle difference in structure (
Fig. 7
); one hypothesis to explain this is that the altered position of the chloride alters the distribution of charge in its ring with respect to the nitrogen atom, and this in turn affects the redox potential of the parabenzoquinone. A dose response for compound 5-1 showed activity in C2C12 cells in the nanomolar range, with significant toxicity in the micromolar range (
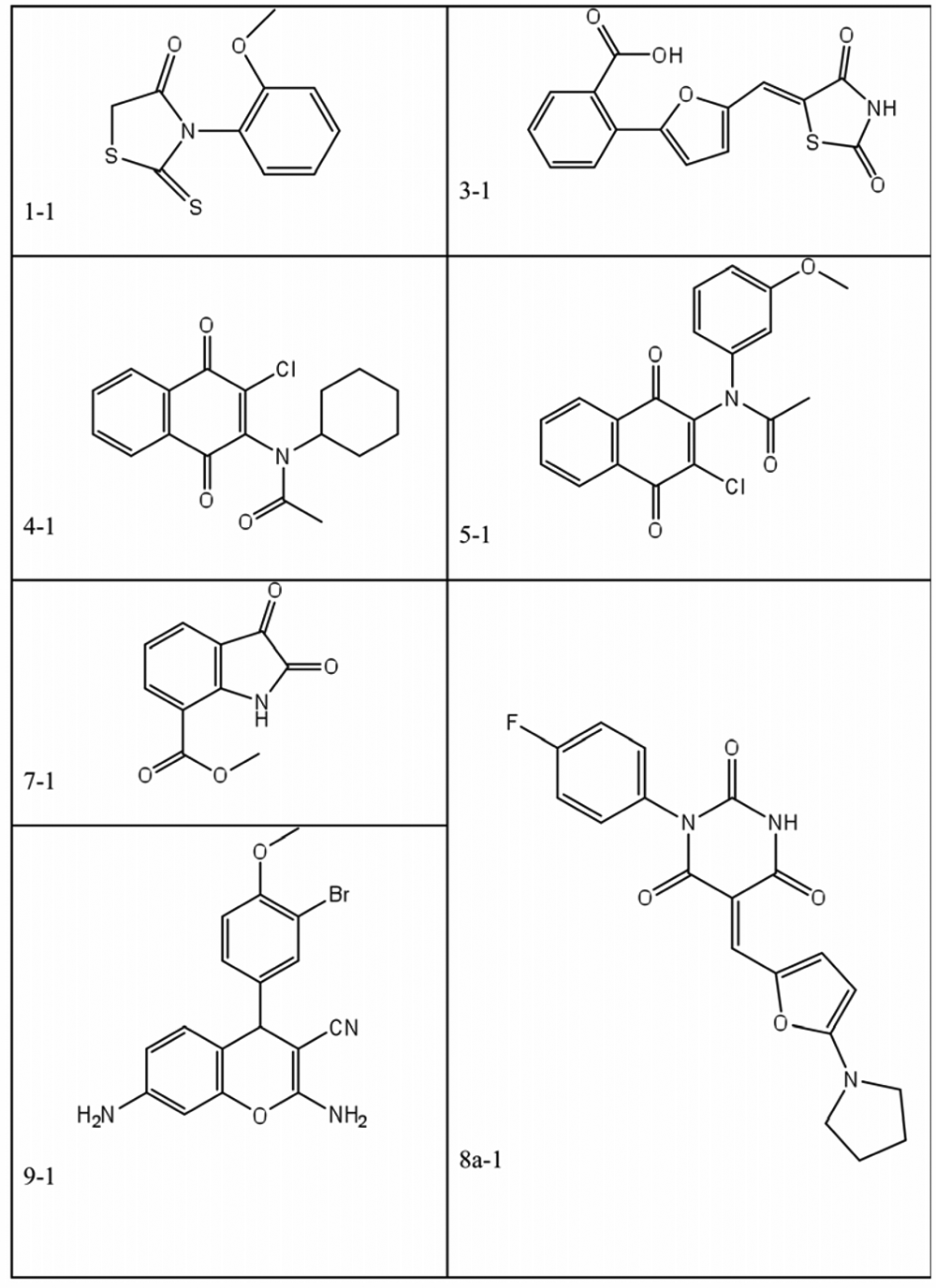
Representative compounds from various core structure groups shown in Figure 4 . Compounds 1-1, 3-1, 4-1, 5-1, 7-1, 8a-1, and 9-1 were identified in the primary high-throughput screening.
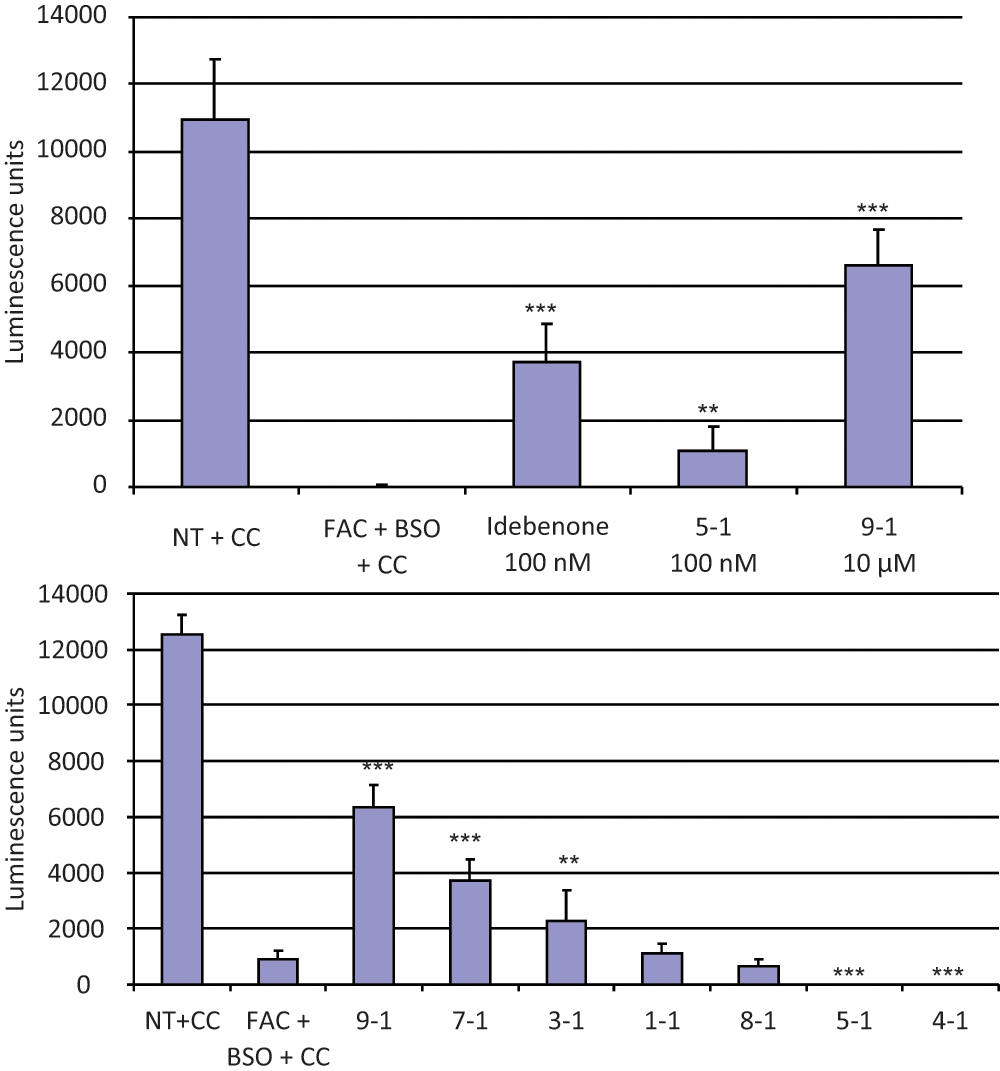
Secondary screening in C2C12 cells of representative compounds (
Fig. 5
) from the various core-structure groups (
Fig. 4
). C2C12 murine myoblast cells were seeded in 96-well plates and treated with a combination of 100 µg/mL ferric ammonium citrate (FAC) and 100 µM L-buthionine (S,R)–sulfoximine (BSO; columns 2–5) for 48 h and 24 h, respectively, as described in the text and in the
Figure 3
legend. Two hours after the addition of BSO, compounds were added as described below. Drug carrier (DMSO) final concentration was 1%. After 24 h, cells were lysed and their viability assessed by chemiluminescence measurement of intracellular adenosine triphosphate (ATP; see Methods). ATP content is shown on the ordinate in (arbitrary) luminescence units. The data shown in each part of the figure are the mean and the standard deviation calculated from eight replicates. The experiments in each part of the figure are representative of at least two independent experiments. CC, carrier control (DMSO at a final concentration of 1%); NT, no treatment. (Top) Relative potency of compound 9-1 (the most efficacious) and compound 5-1 (the most potent) in the secondary screening. Idebenone (column 3), compound 5-1 (column 4), and compound 9-1 (column 5) were added to the final concentrations indicated. Compound 5-1, at its most favorable concentration (
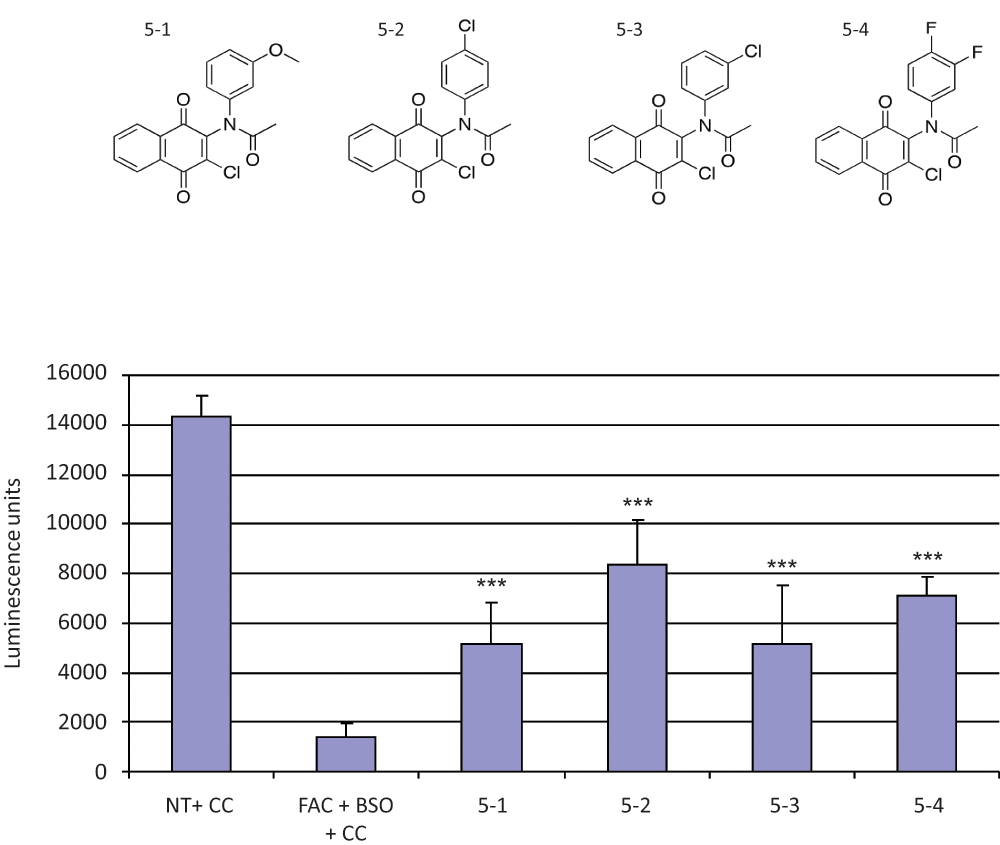
Structure–activity relationships among derivatives of compound 5-1. (Top) Structures of compound 5-1 and analogues 5-2, 5-3, and 5-4. (Bottom) C2C12 murine myoblast cells were seeded in 96-well plates and treated with a combination of 100 µg/mL ferric ammonium citrate (FAC) and 100 µM L-buthionine (S,R)–sulfoximine (BSO; columns 2–6) for 48 h and 24 h, respectively, as described in the text and in the Figure 3 legend. Two hours after the addition of BSO, compounds 5-1, 5-2, 5-3, and 5-4 were added to a final concentration of 100 nM (columns 3, 4, 5, and 6, respectively). Drug carrier (DMSO) final concentration was 1%. After 24 h, cells were lysed and their viability assessed by chemiluminescence measurement of intracellular adenosine triphosphate (ATP; see Methods). ATP content is shown on the ordinate in (arbitrary) luminescence units. All of the compounds partially rescued the C2C12 cells treated with FAC and BSO. In addition, both compound 5-2 and compound 5-4 increased ATP more than compound 5-1 (two-sided p = 0.002 and p = 0.01, respectively, using Student t test). Compound 5-2 increased ATP more than compound 5-3 (two-sided p = 0.008, using Student t test). The data shown in each part of the figure are the mean and the standard deviation calculated from eight replicates. The experiments in each part of the figure are representative of at least two independent experiments. CC, carrier control (DMSO at a final concentration of 1%); NT, no treatment. ***Two-sided p < 0.001 compared with carrier control (using Student t test).
It has been hypothesized that parabenzoquinones, such as CoQ10 and idebenone, help FRDA cells by enhancing the flux of electrons through the mitochondrial ETC, particularly from complex I. Because the compounds identified in our HTS assay are known to be active in the yeast S. cerevisiae, for which there is a complete library of strains in which each gene has been systematically knocked out, we used a novel genetic approach to explore potential mechanisms of action of our hit compounds. We first tested whether compounds from various core-structure groups shown in Figure 4 could increase ATP in yeast lacking Yfh1p. Because homoeostatic mechanisms keep intracellular ATP within narrow limits, we determined the immediate effect of compounds on ATP levels (i.e., within minutes of adding compounds to the yeast cells). Strikingly, we found that the parabenzoquinones in particular induced an immediate burst of ATP synthesis—more than 10-fold—when added to yeast lacking Yfh1p ( Fig. 8A ). This ATP burst required cells to be present and intact—that is, neither compound alone nor prelysed cells (using the CellTiter-Glo reagent) plus compound produced the effect (data not shown). We hypothesize that this ATP burst reflects high intracellular adenosine diphosphate (ADP) and a pent-up demand for ATP.
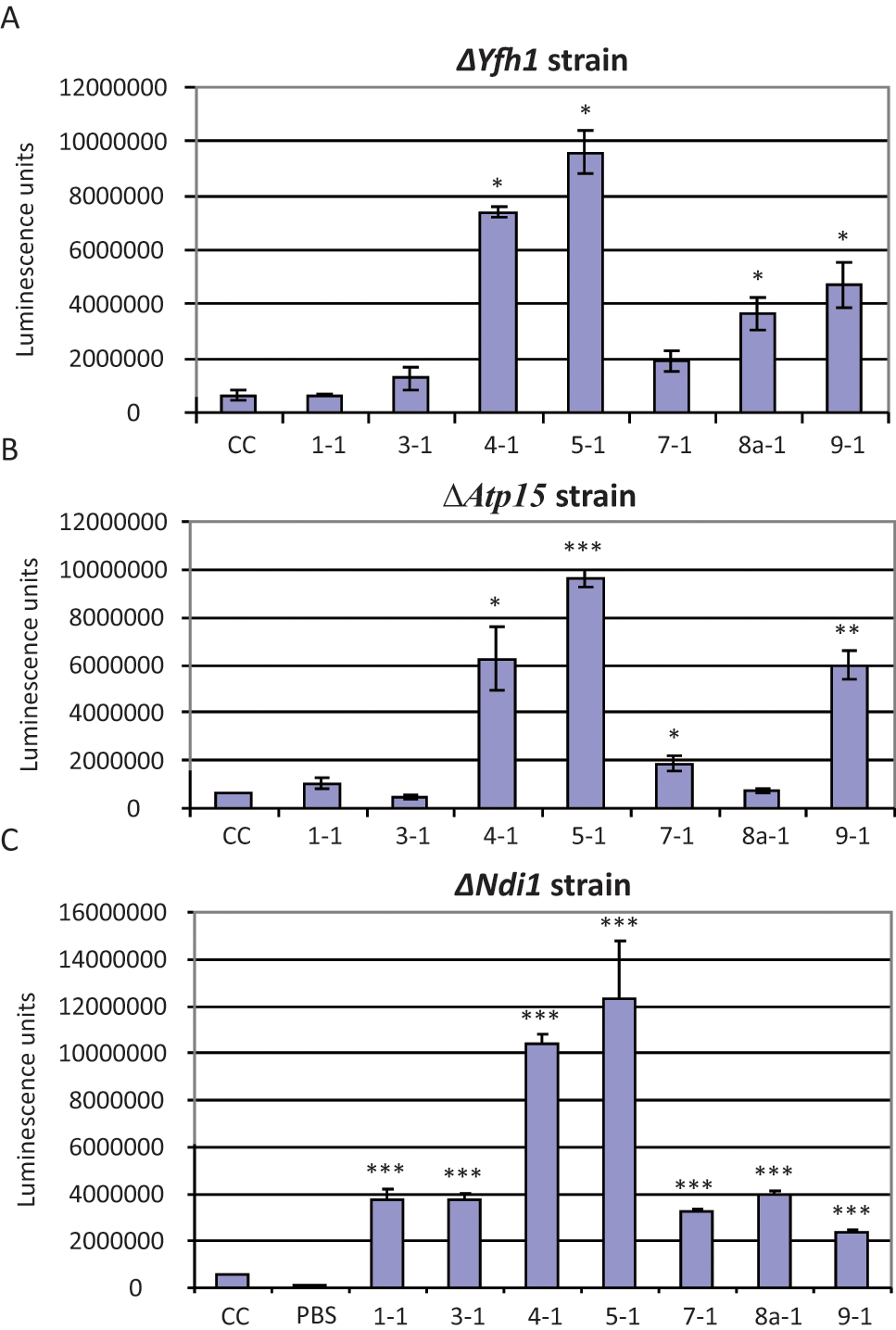
Adenosine triphosphate (ATP) burst assay in yeast knockout mutants. Yeast cells harvested at early log phase (optical density [OD] = 0.2) were washed with water, resuspended in SC-Gly medium, and mixed with either DMSO (CC, carrier control) at a final concentration of 1% (column 1) or with compounds at 100 µM (columns 2–8). Cells were then lysed and ATP content quantified by adding an equal volume of CellTiter-Glo reagent (Promega, Madison, WI). ATP content is shown on the ordinates in (arbitrary) luminescence units. The ATP burst seen with compounds 4-1 and 5-1 is detected in all three yeast strains (columns 4 and 5). The data shown are the mean and the standard deviation of three replicates. The experiments shown are representative of at least two independent sets of experiments. (A) ATP bursts in ΔYfh1 yeast cells treated with the compounds indicated; the bursts seen with compounds 4-1, 5-1, 8a-1, and 9-1 were statistically significant compared with carrier control. (B) ATP bursts in ΔATP15 yeast cells, which lack functional complex V in the electron transport chain, treated with the compounds indicated; the bursts seen with compounds 4-1, 5-1, 7-1, and 9-1 were statistically significant compared with carrier control. (C) ATP bursts in ΔNdi1 yeast cells, which lack functional complex I in the electron transport chain, treated with the compounds indicated; the burst seen with every compound was statistically significant compared with carrier control. PBS, phosphate-buffered saline. *Two-sided p < 0.05 compared with carrier control (using Student t test). **Two-sided p < 0.01 compared with carrier control (using Student t test). ***Two-sided p < 0.001 compared with carrier control (using Student t test).
We then determined whether the ATP burst was lost in a ΔAtp15 yeast strain, which lacks functional complex V in the ETC; surprisingly, the ATP burst associated with the parabenzoquinones was still present, implying that the ATP was not produced from the ETC (
Discussion
We describe herein three novel drug-screening assays for FRDA. The first, a primary HTS assay, takes advantage of Yfh1p, the yeast orthologue of human frataxin, decreased expression of which causes FRDA. Using an inducible-repressible promoter construct, yeast are depleted of Yfh1p, and the consequent mitochondrial dysfunction is quantified by tetrazolium dye reduction. From an initial small pilot study, a series of compounds that rescue mitochondrial function was successfully identified. One of these initial hits was then used as a positive control to optimize the assay for an HTS campaign. In retrospect, the compound used as a control, tannic acid, was suboptimal but was the best available at the time. During the screening campaign, the tannic acid control exhibited significant variability (
More than 100 000 compounds were run in the assay, and 640 hits were identified and screened in follow-up dose–response. Of these, 302 compounds were identified that effectively rescue mitochondrial function, making them excellent candidates for further evaluation. One of these compounds was selected as a positive control for a later HTS campaign (manuscript in preparation), and the new control compound performed consistently throughout the screen. This supports our decision to proceed with the screen described herein despite control metrics that were suboptimal for HTS screening. In cases where there are no known controls or no good control, the only way to find one is to run the screen.
To confirm activities in mammalian cells, a secondary screening assay was developed using the murine myoblast cell line C2C12. The FRDA phenotype was recapitulated by treating the cells with iron and BSO, which inhibits glutathione synthesis. We chose concentrations of iron and BSO that are sublethal by themselves but together produce a synergistic loss of viability, as measured by loss of ATP content. Assessment of hit compounds by rescue of viability in this secondary assay demonstrated a good correspondence between activity in yeast and activity in mammalian cells.
To begin to understand mechanisms of action, we developed a novel genetic approach. We took advantage of the yeast model system, in which (a) our compounds were already known to be active, (b) the essential components of mitochondrial bioenergetics are evolutionarily conserved, and (c) knockout strains for essentially all known genes are available. Using yeast lacking functional components of specific complexes of the ETC, we showed that the immediate increase in ATP induced by hit compounds in yeast lacking Yfh1p was dependent on the ETC for some compounds but independent of the ETC for other compounds. These results demonstrate how mechanisms of action of hit compounds, particularly those targeted to respiratory chain diseases, might be elucidated by taking advantage of the genetic flexibility of yeast.
In summary, the compounds identified in this study have potential relevance for the treatment of FRDA and for other neurodegenerative disorders associated with mitochondrial dysfunction, such as Parkinson disease.
Footnotes
Acknowledgements
We thank Nice Shindo for technical assistance.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported by the NIH-NINDS High Throughput Drug Screening Facility for Neurodegenerative Disease Program (contract N01-NS-22348; Dr. Jill Heemskerk, Project Officer), by R21 grants (NS045331 and NS053546) to R.B.W. from the NINDS, and by grants to R.B.W. from the Institute on Aging of the University of Pennsylvania, the Friedreich’s Ataxia Research Alliance, and the Hartford Foundation.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.