
Abstract
Objectives:
The rare inherited autosomal recessive disease Friedreich ataxia (FA) causes progressive neurodegenerative changes and disability in patients. A systematic literature review (SLR) was carried out to understand and summarize the published efficacy and safety of therapeutic interventions in this disease.
Methods:
Database searches were carried out in MEDLINE, Embase, and Cochrane by two independent reviewers. In addition, trial registries and conference proceedings were hand-searched.
Results:
Thirty-two publications were deemed eligible according to PICOS criteria. Twenty-four publications detail randomized controlled trials. The most frequently identified therapeutic intervention was idebenone (n = 11), followed by recombinant erythropoietin (n = 6), omaveloxolone (n = 3), and amantadine hydrochloride (n = 2). Other therapeutic interventions were investigated in one publication: A0001, CoQ10, creatine, deferiprone, interferon-γ-1b, the L-carnitine levorotatory form of 5-hydroxytryptophan, luvadaxistat, resveratrol, RT001, and vatiquinone (EPI-743). These studies included patients from 8 to 73 years old, and disease duration varied from 4.7 to 19 years. Disease severity as per the mean GAA1 and GAA2 allele repeat length ranged from 350 to 930 and 620 to 987 nucleotides, respectively. Most frequently reported efficacy outcomes were the International Cooperative Ataxia Rating Scale (ICARS, n = 10), the Friedreich Ataxia Rating Scale (modified FARS and FARS-neuro, n = 12), the Scale for Assessment and Rating of Ataxia (SARA, n = 7), and the Activities of Daily Living scale (ADL, n = 8). Each of these assesses the severity of disability in FA patients. In many studies, patients with FA deteriorated according to these severity scales regardless of treatment, or inconclusive results were found. Generally, these therapeutic interventions were well-tolerated and safe. Serious adverse events were atrial fibrillation (n = 1), craniocerebral injury (n = 1), and ventricular tachycardia (n = 1).
Conclusion:
Identified literature showed a considerable unmet need for therapeutic interventions that halt or slow the deteriorating nature of FA. Novel efficacious drugs should be investigated that aim to improve symptoms or slow disease progression.
Plain Language Summary
Friedreich ataxia (FA) is a rare genetic condition that causes nervous system damage and movement problems, including muscle weakness and impaired coordination (ataxia). Heart problems, vision problems, spine problems, and diabetes can occur, too. Within 10 to 20 years of the first symptoms, an individual with FA generally requires a wheelchair.
Currently there are no approved treatments for FA. Current treatments focus on relieving symptoms. This study was carried out to obtain a landscape view of all the published evidence about FA treatments.
• Two scales were most frequently used to assess disease severity: the International Cooperative Ataxia Rating Scale (ICARS) and the FA Rating Scale (modified FARS and FARS-neuro).
• Patients on idebenone at 1350 to 2550 mg per day showed improvement in ICARS and FARS scores over 6 months, but scores deteriorated after 12 months in ambulatory patients with FA.
• Omaveloxolone at doses of 2.5 to 300 mg per day showed significant improvement in mFARS scores and FA Activity of Daily Living scores at 48 weeks compared with placebo.
• Patients treated with vatiquinone showed significant improvements in FARS-neuro scores at 24 months versus natural disease progression.
• Other treatments did not show evidence of significant improvement.
FA leads to nervous system damage slowly, over an extended period. It is important to keep in mind that many of the studies reviewed here were of fairly short duration, meaning that the effects of a treatment may not have been detectable.
This study was undertaken in the hopes that a comprehensive picture of the current treatment landscape for FA will help promote research that will eventually lead to effective treatments to slow down or reverse the damage caused by disease, which are vitally needed.
Introduction
Friedreich ataxia (FA) is a rare inherited autosomal recessive disease that causes progressive neurodegenerative changes and disability due to impaired muscle coordination. 1 It is caused by the repeat expansion of GAA trinucleotide owing to the mutations in the FXN gene, which further impairs the transcription of frataxin protein. 2 A decrease in frataxin concentration leads to mitochondrial cellular dysfunction and disruption in the activity of iron–sulfur cluster biosynthesis, 2 which causes the phenotypic characteristics of the disease. FA is characterized by clinical features including limb ataxia, impaired muscle coordination, cardiomyopathy, gait disturbances, scoliosis, fatigue, visual impairment, and proprioceptive loss. About 10–30% of FA patients experience diabetes.3,4 FA is the most common form of hereditary ataxia, mostly prevalent in Europe (about 1 in 50,000); there are about 1.7 cases per 100,000 to 4 cases per 100,000 in the United States, Middle East, and South Asia.3,5–7
The age of disease onset generally ranges from 5 to 15 years, with most individuals experiencing wheelchair dependence within 15 years of diagnosis. 2 Neurological symptoms such as limb and gait ataxia, loss of reflexes, and dysarthria degrade as the disease progresses, and patients experience pyramidal muscle weakness and loss of ambulation, both of which severely affect the activities of their daily life. As the disease progresses, cardiac complications such as cardiomyopathy and arrhythmia cause mortality in about 60% of patients.3,4
Normal alleles in the FXN gene contain fewer than 33 GAA repeats, whereas FA patients have abnormal GAA repeat expansion of 66 to 1500. The length of the GAA repeat expansion is inversely correlated with the age of onset and the onset of cardiomyopathy and directly correlated with disease severity. 8 The average lifespan of FA patients is about 36 to 40 years from the onset of disease. 9
Evidence also suggests that quality of life (QoL) in FA patients is severely impacted by psychological and physical symptoms. An Australian study of 63 patients found that QoL of FA patients as measured in all areas of the SF-36, a tool that assesses patients’ mental and physical QoL, was significantly worse than that of the general Australian population 10 and that dimensions such as physical functioning and general health were impacted the most. In patients with severe disease, the domain of physical functioning was most impacted. 10
Existing treatment strategies are multidisciplinary and mostly focus on symptomatic treatment of the disease. 6 Consensus clinical guidelines (2014) 11 and Ataxia UK (2016 and 2019)12,13 recommend using physical therapy and aerobic training to treat gait disturbances, and speech and language therapy to treat dysarthria.11–13 Neuropathic pain is treated by anti-neuropathic pain medications such as gabapentin, pregabalin, lamotrigine, and antidepressants. Heart complications that occur in FA are treated with medications such as angiotensin-converting enzyme inhibitors (e.g. enalapril, ramipril), angiotensin 2 receptor blockers (e.g. candesartan, valsartan), and beta blockers (e.g. carvedilol, bisoprolol, metoprolol). Surgery is recommended in severe cases of scoliosis.11–13 Problems related to progressive dysarthria and dysphagia require specialist care and monitoring. Pharmacological treatments (baclofen and botulinum toxin) and stretching programs aid in countering spasticity; insulin is usually given to FA patients with diabetes. Psychological counseling is also given to patients in many cases. Rehabilitation therapy aids functional movements in patients with ataxia and weakness. Regular neurological and cardiological examinations are recommended to monitor the progression of the disease.11–13 Although the disease symptoms improve in some patients with these therapies, QoL significantly worsens with disease progression, especially in patients with early disease onset. 5
Antioxidant therapies such as idebenone, coenzymeQ10, and vitamin E have been assessed for the treatment of patients with FA. A review by Kearney et al. 14 that collated and presented evidence on the efficacy of antioxidants used for the treatment of patients with FA reported inconclusive evidence regarding the effectiveness of antioxidants such as idebenone, vitamin E, coenzyme Q10 on the neurological features of FA; however, other therapies such as deferiprone (iron chelator), interferon gamma-1b, and erythropoietin (EPO) were not included in the review.
There is an unmet need in treating the underlying disease, owing to which the treatment landscape of FA is evolving, with many molecules in the development pipeline. Several therapies are currently being investigated in clinical trials targeting different pathological processes associated with FA that may alter the progression of the disease on a cellular level. 15 This may slow disease progression and benefit patients by improving their QoL. Drugs such as omaveloxolone, vatiquinone (EPI-743), RT001, NAD+ (along with exercise), and MIB-626 aim to improve mitochondrial function and reduce the oxidative stress associated with FA, whereas MIN-102, IMF, and dimethyl fumarate aim to modulate frataxin-controlled metabolic pathways. Some molecules are being developed as a frataxin stabilizer, enhancer, or replacer such as CTI-1601, etravirine, and EPO. Some others, such as resveratrol, gene TACs, and oligonucleotides, aim to increase FA gene expression. Immune modulators that aim to increase FXN expression such as IFNγ-1b are also under development. Gene replacement therapy is another promising approach, now being tested as a treatment to reverse the symptoms associated with the disease.15–17
Given that many products are currently under development, a comprehensive picture of the current treatment landscape for FA will help promote research that might eventually lead to effective treatments, which are vitally needed. Hence the aim of this research was to systematically identify and summarize published literature on the efficacy, effectiveness, and safety of therapeutic interventions in FA.
Methods
A systematic literature review (SLR) was carried out according to requirements set by the Cochrane Handbook and the Centre for Reviews and Dissemination (CRD), 18 as well as those contained in the 27-item PRISMA checklist. 19 This SLR aimed to include English publications that summarized trial results regarding the efficacy, effectiveness, and safety of different interventions in FA.
To find these publications, a search strategy was developed, after which electronic searches in the databases of MEDLINE (In-Process), Embase, and the Cochrane Library were carried out on 16 July 2021. In addition, conference searches were conducted covering proceedings from the last 3 years (2019–2021) of the Muscular Dystrophy Association Annual Meetings, the Clinical and Scientific Conference, the Friedreich’s Ataxia Research Alliance (FARA) Symposium, the Cognitive Neuroscience Society (CNS) Annual Meeting, the American Academy of Neurology (AAN) Annual Meeting, the European Academy of Neurology (EAN) Annual Meeting, the European Conference on Rare Diseases and Orphan Products, and the Rare Diseases and Orphan Products Breakthrough Summit. Furthermore, searches in clinical trial registries (clinicaltrials.gov, EU Clinical Trials Register, and the World Health Organization’s International Clinical Trials Registry Platform [WHO-ICTRP]) were performed.
Search strategy
The search strategy included a combination of keywords pertaining to the population of interest and study design. The search terms included medical subject headings (MeSH) and Embase subject headings (Emtree) terms, and keywords. Relevant articles were identified by a search of the titles and abstracts of peer-reviewed publications. Search terms for randomized controlled trials (RCTs) and observational studies were based on the filters provided by the Scottish Intercollegiate Guidelines Network (SIGN). 20 The detailed search strategy can be found in the Supplementary Tables S2 to S4.
Selection of studies
Eligibility criteria were derived using population, intervention, comparator, outcomes, and study design (PICOS) criteria. Publications assessing the effectiveness or efficacy and/or safety of interventions in patients with FA at any age and stage in a randomized or non-randomized study were included. Eligibility criteria can be found in the Supplementary Table S1.
Two reviewers independently screened the title and abstracts of the articles in the search results and excluded those that did not meet the eligibility criteria. In the case of a conflict, a third reviewer made the final decision. The same process was applied at the full-text screening stage. During both the title and abstract and the full-text screening stages, reasons for exclusion of articles were documented. The quality of the included RCTs and non-RCTs was assessed using the CRD appraisal checklist. 18
Note that the intent of this SLR is not to provide an explicit comparison between the studies identified but to provide a summarized qualitative review of efficacy of interventions assessed in FA.
Results
Included studies and PRISMA flow diagram
Overall, 1859 hits were retrieved from electronic databases. After deduplication, 1271 publications were screened at the title and abstract stage. Subsequently, 189 hits were screened for eligibility during the full-text screening stage, of which 157 hits were excluded for the following reasons: intervention (n = 7), non-relevant comparator (n = 31), and study design (n = 106). A total of 32 publications were included for data extraction and quality assessment. In addition, a total of 104 hits were obtained via the gray literature hand searches. None of the citations met the eligibility criteria and were therefore excluded. A PRISMA flow chart detailing the different parts of the screening process can be found in Figure 1. The PRISMA reporting checklist is available in Supplementary materials.

PRISMA flowchart.
Study characteristics
A total of 322,4,21–50 publications were identified, of which 282,4,22–26,28–37,39–49 were unique studies. Overall, 22 RCTs2,4,21,23,25,26,28,31–36,38,40–43,46–50, 5 open-label studies,23,27,30,40,44 2 single-arm trials,37,39 2 observational studies,24,29 and 1 crossover study 45 (Table 1) were reviewed. Of the two observational studies, one was prospective 24 in nature and the other retrospective. 29 The majority of the studies were conducted in the United Kingdom (n = 18), the United States of America (n = 12), Australia (n = 5), and Canada (n = 2). Study duration ranged from 2 weeks 28 to 7 years, 44 and sample size ranged from 5 39 to 104 44 patients (Table 1).
Study and patient characteristics.

^, indicates frequency (%); *, indicates median value CoQ10, coenzyme Q10; IQR, interquartile range; IU, international units; LVH, left ventricular hypertrophy; Mo, months; RhuEPO, recombinant human erythropoietin; SD, standard deviation; SE, standard error; SEM, standard error of the mean; UK, United Kingdom; US, United States; Wk, weeks; Yrs, years.
Treatment and patient characteristics
The patient population inclusion criteria varied among these studies, but the majority included genetically confirmed patients with FA. Most studies excluded patients with comorbidities such as diabetes, hypertension, and cardiac disease, as well as pregnant and breastfeeding women. Idebenone was the most frequently assessed therapy (n = 11), followed by recombinant EPO (n = 6), omaveloxolone (n = 3), amantadine hydrochloride (n = 2); and A0001, CoQ10, creatine, deferiprone, interferon-γ-1b, L-carnitine levorotatory form of 5-hydroxytryptophan, luvadaxistat, resveratrol, RT001, and vatiquinone (all n = 1). The mean age of study participants in these trials ranged from 12.7 36 to 40 years, 37 and mean disease duration ranged from 4.7 2 to 19 40 years. The percentage of male participants varied between 0% 22 and 85%. 37 The mean GAA1 and GAA2 allele repeat length, an indication of FA severity, ranged from 350 39 to 930 47 and 620 50 to 987 36 nucleotides, respectively.
Different instruments have been used to map the severity of the underlying symptoms associated with FA. Instruments such as the Scale for Assessment and Rating of Ataxia (SARA), the International Cooperative Ataxia Rating Scale (ICARS), and the (modified) FA Rating Scale (mFARS) have been designed to underscore the impairment associated with the neurological and ataxia-related progression in these patients. The Activities of Daily Living scale (ADL) is a component of the FARS scale and gives insight into how well patients can perform their activities each day, whereas FARS-neuro is a neurological component of the FARS. The baseline severity of patients was reported in 23 of 32 publications. An overview of the baseline severity of patients in the included publications can be found in Table 2. Baseline FARS scores ranged from 47.5 to 98.5 (0–159), ADL scores from 9.5 to 11.5 (0–36), ICARS scores from 31 to 58.8 (0–100), and SARA scores from 16.3 to 24.1 (0–40). Generally, higher scores indicate greater disability.
Baseline severity of disease in FA patients.

ADL, activities of daily living; CEPO: carbamylated erythropoietin; CoQ10: coenzyme Q10; EPO: erythropoietin; FA, Friedreich ataxia; FARS, Friedreich Ataxia Rating Scale; HC, health controls; IQR, Interquartile range; ICARS, International Cooperative Ataxia Rating Scale; mFARS, modified Friedreich Ataxia Rating Scale; Med, Median; rhuEPO: recombinant human erythropoietin; SARA, Scale for Assessment and Rating of Ataxia.
Efficacy outcomes
The included publications are heterogenous in their study and patient characteristics, duration of follow-up, and outcome measures. The treatments also vary in their mechanism of action. Hence, a quantitative estimation of effects or a meta-analysis of publications cannot be performed. Moreover, many publications have a small sample size, limiting their generalizability. We, therefore, provide a qualitative overview of the clinical evidence of interventions in FA segregated by randomized and non-randomized studies.
FARS scale
FARS scores (including mFARS and FARS-neuro) were reported in 12 publications2,4,26–28,32,40–43,48,50 with the aim to measure the efficacy of interventions on neurological and ataxia-related progression in FA. FARS scale scores range from 0 to 159, with higher scores signaling greater disability in FA patients. The modified FARS scale is a widely used FA-specific scale for assessing the efficacy of the intervention in clinical trials and was reported in four publications.2,4,48,50 The FARS-neuro was independently administered in two studies.26,32 Changes from baseline scores for FARS, mFARS, and FARS-neuro scales for all publications are presented in Figures 2–4, respectively.

Mean change from baseline in FARS scores.
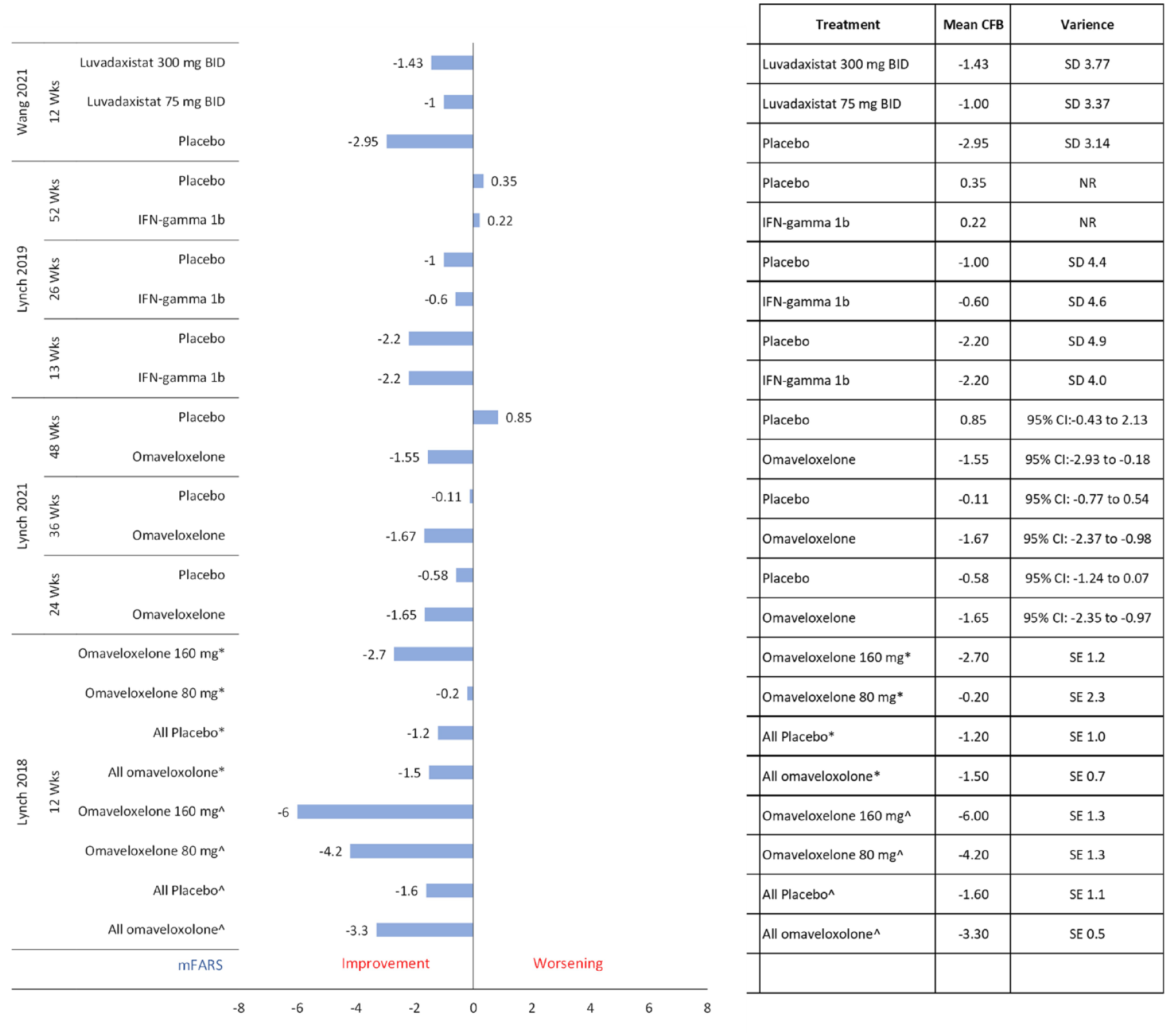
Mean change from baseline in mFARS scores.

Mean change from baseline in FARS-neuro scores.
In randomized studies, mean FARS scores improved slightly with both idebenone high dose (1350 or 2250 mg) and low dose (450 or 900 mg) in a 6-month IONIA study, 43 but the scores eventually worsened in an extension study (IONIA-E) 27 with idebenone at 12 months and at 18 months; in this study, patients in the high-dose arm (1350 or 2250 mg/kg) continued with their treatment for another 12 months. Worsening FARS scores signaled deterioration of patients’ ataxia on treatment with high doses of deferiprone (40 and 60 mg/kg) in the study by Pandolfo et al. 41 No change in mean baseline FARS scores was observed with carbamylated EPO. 28 Treatment with IFN-gamma 1b showed moderate levels of improvement in mean FARS scores, but greater improvements in mean FARS scores were observed among patients in the placebo arm, potentially indicating a placebo effect. 48 A dose-dependent, statistically significant improvement in mean FARS score was observed in patients receiving treatment with A0001 0.5 and 0.75 g 42 as compared with placebo.
Mean mFARS scores consistently improved with both omaveloxolone 160 and 80 mg treatment in a dose-dependent manner at 12 weeks as reported by Lynch et al. 50 in a dose-ranging RCT. It was observed that patients with pes cavus (high foot arch) had less improvement in mFARS scores with omaveloxolone than patients without pes cavus. In addition, the higher dose of omaveloxolone was examined in a larger, phase 3 trial, where patients treated with omaveloxolone 150 mg showed a steady improvement in baseline mFARS scores over 48 weeks. 2 Patients treated with IFN-gamma-1b showed improvement after 13 weeks of treatment, but their mFARS scores thereafter gradually deteriorated during a 52-week study. 48 Also, the improvement in the placebo arm was greater than that in the intervention arm at 13 and 26 weeks. This observation echoed the findings of change from baseline in FARS scores, with IFN-gamma-1b 48 further denoting a placebo effect. Similarly, in a phase 2 trial of luvadaxistat, mFARS score improvement was higher among patients in the placebo group than among patients in the intervention group. 4
A short 6-month study of vatiquinone at two doses (200 and 400 mg) was not able to demonstrate statistically significant differences in the FARS-neuro scores. However, patients were treated in an open-label extension where a decrease in FARS-neuro scores of 1.8 units was noted at 24 months. This improvement in FARS-neuro was significantly different than the worsening in scores in a natural history cohort from the FA Common Outcome Measure Study (p < 0.001) which was matched on factors such as age, sex, and FARS-neuro score. 32 Similarly, improvement in FARS-neuro scores was observed in the phase 1/2 trial for RT001, but statistical significance was not reached versus placebo. 26
Only one non-randomized study reported scores relating to FARS. Resveratrol in an open-label trial demonstrated a dose-dependent improvement, that is, a greater reduction of mean FARS scores from baseline for 5 g dose when compared with 1 g dose. 40
ADL scale
The ADL scale is a component of the FARS scale. The ADL scale assesses how well FA patients can carry out their daily activities. It ranges from 0 to 36, in which higher scores signal greater disability. In total, eight publications reported ADL scores. The mean change from the baseline scores of the ADL scale in the eight publications is presented in Figure 5.

Mean change from baseline in ADL scores.
In randomized studies investigating deferiprone, 41 coenzyme Q10, 25 idebenone, 43 and IFN-gamma-1b, 48 mean ADL scores deteriorated over time, whereas treatment with EPO showed negligible improvement in the scores in one publication. 34 Similarly, treatment with omaveloxolone resulted in improved scores as compared with placebo in a 48-week study, 2 although the improvement from baseline was minor and statistically non-significant. Low-dose A0001 42 and luvadaxistat 75 and 300 mg 4 both showed improved ADL scores; however, the differences were not statistically significant versus placebo.
No non-randomized study reported ADL scores.
ICARS scale
The ICARS scale quantifies the level of impairment caused by hereditary ataxia such as FA. Ten publications reported its use in measuring efficacy outcomes.1,24,25,27,30,33,40,41,43,44 Three of these publications25,27,44 additionally reported ICARS subscores for eye, speech, upper limb, lower limb, posture, and stability and spiral subscores. ICARS scores range from 0 to 100, with higher scores indicating greater disability. The mean changes from baseline for ICARS scores from the 10 publications are presented in Figure 6.

Mean change from baseline in ICARS scores.
There is conflicting evidence from the publications assessing the effectiveness of idebenone treatment. In randomized studies such as the IONIA study by Lynch et al., 43 greater improvements in mean ICARS scores were seen with placebo at 12 weeks as compared with idebenone, but at 24 weeks, patients receiving treatment with idebenone reported greater improvement from baseline than those receiving placebo, indicating a delayed effect. However, data from the IONIA-E, an extension study of 12 months, concluded that a higher dose of idebenone (1350 mg/kg) slightly worsened the mean ICARS scores at 12 months, although pooled analysis showed improved scored from the baseline of the IONIA study to 12 months (52 weeks) of the extension study. 27 Alternatively, in a 2-month study, 23 idebenone significantly improved mean ICARS scores but only in ambulatory patients.
Worsening of scores was also observed in the study by Pandolfo et al., 41 where high doses of deferiprone (40 and 60 mg/kg) worsened patients’ ataxia indicated by an increase in mean ICARS scores, which eventually lead to the discontinuation of the 60 mg/kg arm in the study. Slight non-significant improvement was also seen with creatine as compared with placebo in a study by Schöls et al. 33
In non-randomized studies, a similar trend of worsening of mean ICARS scores with long-term idebenone treatment was observed. Arnold et al. 24 also reported worsening of mean ICARS scores with idebenone at ~3 years. In addition, idebenone worsened ataxia of patients in a long-term follow-up study of 7 years as reported by Ribaï et al. 44 and interestingly the worsening was higher in the idebenone group as compared with the placebo group. Hence, evidence from both randomized and non-randomized studies point out the inefficacy of long-term treatment with idebenone for improving ICARS scores.
Improvement in mean ICARS scores was observed in only one publication. 40 Resveratrol showed a significant improvement in ICARS scores 40 in 12 weeks of treatment. Treatment with a low-dose of resveratrol (1 g) improved the mean ICARS scores by −0.3 units, whereas treatment with high-dose (5 g) of resveratrol improved mean ICARS scores by −1.9 units, indicating a statistically significant dose-dependent improvement. But the efficacy of resveratrol remains ambiguous since the drug has not been further studied in a large, randomized trial.
SARA scale
SARA is another measure that assesses the disability of patients with FA. Scores range from 0 to 40, with higher scores associated with greater disability. SARA scores were reported in seven publications.22,28,34,37,39,40,47 This scale was most frequently reported in studies assessing the impact of different forms of EPO, that is, recombinant and carbamylated. Figure 7 presents the mean change from baseline in SARA scores in the seven publications.

Mean change from baseline in SARA scores.
None of the randomized studies reported a significant improvement in SARA scores as compared with placebo.28,34,47 Saccà et al. 34 observed a slight decrease in scores with epoetin alpha at week 24 and week 48 as compared with placebo, although the difference was not statistically significant. Similarly, in the study by Boesch et al., 28 patients from both carbamylated EPO and placebo groups had a slight decrease in mean SARA scores but were not significantly different.
In non-randomized studies, a publication by Nachbauer et al. 37 reported that recombinant human erythropoietin (rhuEPO) significantly decreased SARA scores at 8 weeks as compared with baseline, but the study did not include any comparator group and thus the results may be biased. In another publication by Nachbauer et al. 39 rhuEPO increased mean SARA scores in a 3-month study.
Patients treated with a triple therapy of darbepoetin alpha, idebenone, and riboflavin showed a slight improvement in the annual worsening rate of the disease during the first 2 years (−1.42 ± 0.51), but the SARA scores increased in the third year, indicating disease progression. 22
Other measures of FA severity
Other clinical measures identified in these trials were related to cardiological complications such as cardiomyopathy, left ventricular hypertrophy, and arrhythmia, as measured by left ventricular mass index (LVMi), interventricular septal thickness at diastole (IVSTd), and ejection fraction. Another endpoint of interest was the change in frataxin levels, as this might correlate with reduced disease progression.28,39,47 Evidence on patients’ QoL as measured by EQ-VAS (n = 1) 34 and SF-36 (n = 2)22,50 indicates little meaningful improvement in the physical and mental health of the patients receiving active treatment when compared with patients receiving treatment with placebo (results not reported here in this article).
Safety results
Twenty out of 32 publications reported at least one adverse event (AE).2,4,23–26,28,30–32,34,35,40–43,46–48,50 Most reported AEs in the study were nausea (n = 9), headache (n = 7), diarrhea (n = 6), and abdominal pain (n = 5). A high proportion of AEs related to the gastrointestinal tract were reported with high-dose resveratrol, high-dose idebenone, and omaveloxolone. Ataxia-related AEs were most reported with high doses of deferiprone (40 and 60 mg/kg), which resulted in discontinuation of the study arm. 41 Fluctuations in blood-, kidney-, and liver-related lab parameters such as alanine aminotransferase increase, aspartate aminotransferase increase, decreased serum ferritin, decreased white blood cell (WBC) count, chromaturia, and maximum ALT elevations were commonly observed with omaveloxolone,2,50 deferiprone, 41 and resveratrol. 40 Omaveloxolone also showed ALT and AST increases that may be related to the pharmacological activity of the drug. 2 Treatment-emergent AEs that were related to the study drug were observed in about 65% of the patients in the CEPO arm 28 and 55% patients in the luvadaxistat arm. 4
Other drugs such as vatiquinone, CoQ10, A0001, amantadine hydrochloride, EPO, rhuEPO, RT001, and IFN-gamma 1b have reported fewer AEs.
Serious adverse events (SAEs) including atrial fibrillation, sinus and ventricular tachycardia, anemia, palpitations, craniocerebral injury, and idiopathic thrombocytopenic purpura were reported in four publications.2,4,28,43 The majority of SAEs were reported with omaveloxolone. 2
Risk of bias assessment
The CRD checklist determines the quality of the RCTs in domains of selection bias, performance bias, attrition bias, detection bias, and reporting bias. Only 426,34,43,48 out of the 22 RCTs had a good quality of conduct and reporting as assessed by the domains of the CRD checklist. These four RCTs had adequately reported the method for generating a random sequence along with adopting adequate methods for allocation concealment, thus preventing selection bias. Studies were also double-blinded, and the groups were comparable at the treatment outset. Furthermore, the studies had reported all outcome measures as described in the ‘Methods’ section along with analyzing participants by intention-to-treat methods. They also used appropriate methods to account for missing data, thus preventing attrition bias.
Further details are presented in Supplementary Tables S6 and S7.
Discussion
The main objective of this SLR was to identify all relevant clinical evidence of interventions in FA. The efficacy results of the included studies generally provided inconclusive evidence related to FARS, ICARS, SARA, and ADL scores associated with the same treatments in different studies and for different time points; for instance, idebenone improved ICARS scores in a 6-month IONIA study 38 and in a study by Cook et al. 23 (only ambulatory patients), but ICARS scores deteriorated in a long-term follow-up study (7 years) of idebenone by Ribaï et al. 44 and in a 12-month IONIA-E study. 27 In the case of FARS, scores improved on treatment with IFN-gamma 1b in 13 weeks 48 and with idebenone in a 6-month IONIA study. 38 However, results were not significant as compared with placebo, and moreover, scores worsened with IFN-gamma at 26 weeks 48 and with idebenone in a 12-month IONIA-E study. 27 mFARS scores improved slightly with IFN-gamma 1b after 13 weeks but deteriorated after 52 weeks of treatment. 48 These results indicate that some interventions have slowed FA progression after short-term treatment (2 weeks to 6 months) but failed to demonstrate efficacy in the long term (>6 months). Conversely, omaveloxolone has demonstrated long-term efficacy by significantly improving mFARS scores as compared with placebo in studies up to 48 weeks,2,50 but a substantial number of AEs and SAEs were reported with the drug. FARS-neuro scores did not improve significantly with vatiquinone as compared with placebo in a 6-month trial, but a significant improvement was observed at 24 months as compared with a natural history cohort. 32 This observation could be influenced by the short placebo-controlled phase of this trial. FA-ADL scores also reiterate the same observations as FARS and ICARS scales.
These results are consistent with a previous review conducted by Kearney et al., 14 which concluded that there were no significant differences in ICARS and FARS scores of patients among the assessed intervention and comparator arms. An earlier SLR by Kearney et al. included only two randomized studies and excluded others due to short follow-up (<1 year). They found many studies of low-quality evidence that were deemed insufficient to support or disprove the effect of antioxidants on the progression of FA. Nutraceutical antioxidants such as idebenone and coenzyme Q10 plus vitamin E have been evaluated in earlier trials and most recently in 2019 by Cook et al. 23 owing to their roles in targeting the oxidative stress and iron accumulation in FA. As assessed in this SLR and the previous SLR of the study by Kearny et al., 14 they have provided an inconclusive benefit for FA patients. Both CoQ10 and idebenone in either high or low doses failed to significantly improve ICARS scores from baseline. 25 Idebenone has shown some improvement in the reduction of cardiac hypertrophy, 46 but overall, there was limited effect on the neurological progression of the disease. The definitive answers to the effectiveness of these therapies have remained elusive in multiple trials due to potential defects in study design or the inefficiency of the drug itself, which renders them incomparable to most contemporary trials targeting other pathological pathways of FA. 51
Although this review included 22 articles about randomized trials, of which 11 publications reported on treatment with antioxidants, 4 on treatment with frataxin enhancers, and 3 assessed NRF2 activators. In line with Kearney et al., 14 most therapies failed to demonstrate conclusive and consistent clinical benefit in terms of FARS, ICARS, SARA, or ADL scores.
Based on the current body of evidence, it seems that there is a clear unmet need to find therapeutic interventions that show a significant long-term treatment effect. Although current inconclusive results can be explained by the lack of an efficient drug, many of the studies were conducted with a limited sample size, small separate cohorts for each treatment arm, and short follow-up duration (2 weeks to 6 months), leading to low external generalizability and statistical uncertainty. This necessitates overinterpretation of efficacy results, due to low-powered statistical analyses and difficulties in the ability to assess efficacy outcomes sufficiently and adequately. FA is a heterogenous genetic disease with a variable and slow clinical progression; therefore, treatment effects may not be detectable in short-term trials. More robust trials with longer treatment duration might be needed to determine the safety and efficacy of interventions in FA but might not be possible given the rarity of the disease. Multinational collaboration, engagement of patient groups, and recruitment of patients from different countries might facilitate trials with greater precision and less uncertainty around efficacy results.52,53 Alternatively, using natural history cohorts that have low systematic error and are adjusted for specific characteristics (for example, age, sex, and severity of disease) might support the generation of robust efficacy results in studies where a long-term placebo arm is not feasible. This method was adopted by one of the publications in this SLR 32 that compared an age-, sex- and severity-matched natural history cohort with an intervention cohort at 24 months.
In-depth natural history studies for the disease are recommended to inform sensitive drug development endpoints and required trial duration to conclusively prove a benefit. Another important point is that, at present, there is no validated biomarker available that can act as a surrogate endpoint for indicating a definitive clinical change in the progression of the disease and detect the efficacy of therapies faster than FA-specific scales do. 54 This may aid in further prioritization of therapeutic research and enhanced drug development in this rare disease. Results have been presented side by side, but do not account for heterogeneity between studies in terms of patient characteristics, study design, or dosing regimens. Consequently, the results represent a naïve comparison between studies in terms of FARS, ICARS, SARA, and ADL scores and should be interpreted with caution.
One of the strengths of this review is its systematic approach and its mining of major databases such as Embase and MEDLINE. In addition, the data have been complemented with gray literature from conferences and clinical trial registries. This review used two independent reviewers to mitigate selection bias, in which the reviewers’ perception affects the inclusion of certain studies. This gives the review a certain robustness, as does the fact that it was conducted according to the 27-item PRISMA checklist and CRD guidelines. Considering the rarity of the disease, the reviews included both RCTs and non-RCT publications. However, given the inherent biases of non-RCT studies, their results should be interpreted with caution.
Publication bias might exist, considering that studies that reported less novel or significant results may have been rejected for publication or the results of a trial were not submitted at all. Furthermore, this review considers only publications in English, whereas FA is also prevalent in the Middle East and South Asia. Published literature in other languages that might contain relevant information has not been captured.
Conclusion
In this SLR, none of the investigational therapies had a disease-modifying impact on the natural progression of FA. Although there are indications that a few of these interventions seem to alleviate a few of the symptoms of FA, results often are inconclusive due to small sample size or insufficient follow-up or required a matched natural history cohort or special trial designs, mostly due to studies being underpowered. Hence, there is still an unmet need for evidence that suggests that therapeutic interventions improve the neurological and/or cardiological functions in FA and alter the progression of disease. Clinically robust trials or other techniques are needed to conclusively prove therapeutic efficacy in this rare disease.
Supplemental Material
sj-pdf-1-trd-10.1177_26330040221139872 – Supplemental material for Clinical evidence of interventions assessed in Friedreich ataxia: a systematic review
Supplemental material, sj-pdf-1-trd-10.1177_26330040221139872 for Clinical evidence of interventions assessed in Friedreich ataxia: a systematic review by Paridhi Jain, Lohit Badgujar, Jelle Spoorendonk and Katharina Buesch in Therapeutic Advances in Rare Disease
Footnotes
Acknowledgements
The authors thank Amrita Ostawal for her contributions in reviewing the revised article.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.