
Abstract
High-throughput screening (HTS) efforts for neglected tropical disease (NTD) drug discovery have recently received increased attention because several initiatives have begun to attempt to reduce the deficit in new and clinically acceptable therapies for this spectrum of infectious diseases. HTS primarily uses two basic approaches, cell-based and in vitro target-directed screening. Both of these approaches have problems; for example, cell-based screening does not reveal the target or targets that are hit, whereas in vitro methodologies lack a cellular context. Furthermore, both can be technically challenging, expensive, and difficult to miniaturize for ultra-HTS [(u)HTS]. The application of yeast-based systems may overcome some of these problems and offer a cost-effective platform for target-directed screening within a eukaryotic cell context. Here, we review the advantages and limitations of the technologies that may be used in yeast cell–based, target-directed screening protocols, and we discuss how these are beginning to be used in NTD drug discovery.
Introduction
Neglected tropical diseases (NTDs) are caused by a diverse group of 17 infectious agents: 2 viruses, 4 bacteria, 3 parasitic protozoa (Leishmania and Trypanosoma species), and 8 parasitic helminths (nematodes, cestodes, and trematodes). 1 As diseases of extreme poverty, NTDs blight the lives of 2.7 billion people who live on less than US$2.00/day. NTDs are endemic in tropical and subtropical regions of the developing world, and Africa, Asia, and Latin America carry 90% of the disease burden. Without effective vaccination regimens, many of the parasitic NTDs lack adequate therapeutics, and the few available drugs are often costly and difficult to administer, and/or have unacceptable side effects. 1
Following years of underinvestment, NTDs have recently become the focus of an upsurge of interest as evidenced by the London Declaration.2–3 This represented a commitment by both public and private entities to reduce or even eliminate the burden of NTDs by 2020. Key to realizing this ambitious goal is the sharing of knowledge-based assets among sectors and between partners.2,4 For example, NTD drug discovery may be accelerated by using such open-source strategies as sharing compound collections and screening technologies through product development partnerships. 5 High-throughput screening (HTS) using such collections and technologies is a key approach for the discovery of novel drug leads that can be developed into new pharmaceuticals, including those for NTDs.
High-Throughput Screening for NTDs
HTS facilitates the screening of very large numbers of compounds (typically ≥100,000/day) for biological activity, an approach that is augmented by developments in genomics and combinatorial chemistry, which have revolutionized drug discovery. HTS assays are designed to facilitate screening at a number of biological levels, from individual protein targets to uni- and multicellular organisms. Both in vitro biochemical protein target–based6–7 and parasite cell-based 8 screening technologies have been used in NTD drug discovery efforts. The former approach is generally simpler and more specific; however, the latter, although complex, can provide more context. 9 Both of these approaches have limitations and cost implications (Table 1). For example, the in vitro biochemically based assays often used in screening campaigns against specific targets require purified, soluble, and biologically active protein. In addition, for fully automated HTS, the assay must overcome the need for separation of the reaction product while ensuring an easily detectable and quantifiable signal (e.g., radiochemical or fluorescence). Cell-based platforms, in contrast, negate the requirement for a purified target protein and can be genetically engineered to produce a simple and robust measurable readout of cell response compatible with that of automated HTS. 10 Furthermore, in a cell-based platform, activity is measured in a cellular context; for example, any compound must cross the plasma membrane to reach an intracellular target, thus more accurately reflecting the in vivo situation. Also, a well-constructed cell-based assay can offer the possibility of identifying generically cytotoxic compounds and allowing their elimination from the study. Ideally, to most fully reflect the context in which any compound will have to show an effect, cell-based assays should be performed using the target cell or organism of interest. 11 However, both helminth and protozoan causes of NTDs are relatively challenging to genetically manipulate. In addition, they are often expensive and/or difficult to culture compared to model eukaryotes. 12 Despite this, the relative lack of fully validated and tractable targets in these parasites, coupled with the failure of target-based biochemical screening, 13 has led to the recent preponderance of non-target-directed, cell-based phenotypic screens that rely on cell growth as a measurable output.8,14 The functional redundancy found in complex cellular systems can, however, further complicate the search for specific inhibitors using such cell-based HTS assays. 10 Therefore, although cell-based systems clearly provide some significant advantages compared with conventional target-based in vitro approaches, their use applied to parasitic NTDs is limited by their tractability.
Comparison of High-Throughput Screening Strategies.
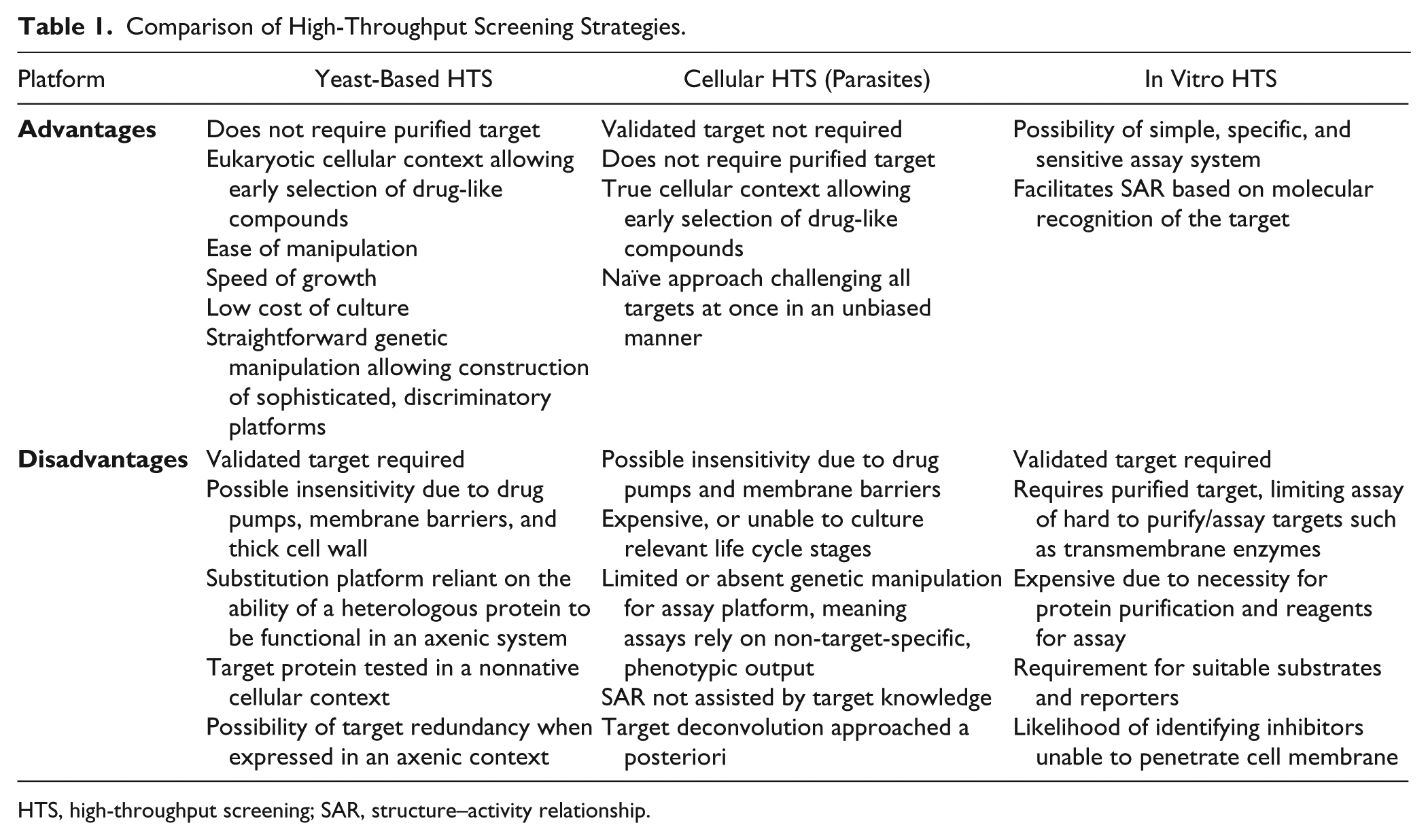
HTS, high-throughput screening; SAR, structure–activity relationship.
Yeast as a Platform for HTS
The yeast Schizosaccharomyces pombe and Saccharomyces cerevisiae represent model and highly tractable organisms, with S. cerevisiae probably being the most highly characterized eukaryote. 15 In addition to genetic tractability, yeast can be grown quickly (doubling time of approximately 90 minutes) in simple, cheap, liquid or solid growth media. These features make yeast ideal vehicles for antifungal discovery. 16 Notably, however, the conservation of cellular functions among yeast, mammalian, protozoan, and other eukaryotes, coupled with the ease of gene knockout, replacement, and overexpression technologies, means that yeast can be engineered to serve as an HTS platform for drug discovery for a range of diseases. 17 Here, we review the possible uses of yeast in HTS for NTD drug discovery, highlighting both the advantages and disadvantages of this platform (Table 1).
Yeast-Based Platforms and Their Relevance to NTD Drug Discovery
Lethal Expression
Exploiting their genetic tractability, yeast can be engineered into reporter systems suitable for HTS. For example, in some instances, the axenic expression of a heterologous protein drug target in yeast results in a measurable phenotypic readout.11,17 This approach has been deployed successfully to identify antiviral agents through screening programs.
Expression of the pro-apoptotic HIV-1 protein Vpr is lethal to the fission yeast S. pombe, a phenotype that has been exploited to format 96- and 384-well yeast-based screens. 18 With Vpr under the control of an inducible promoter and switched on, compounds that lead to growth restoration, presumably via Vpr inhibition, were identified by measuring optical density. This represents a very simple, HTS-compatible, primary assay platform that has been similarly used to identify inhibitors of the influenza virus ion-channel-forming protein in S. cerevisiae. 19 Notably, the Vpr-expressing yeast were also used in a secondary, fluorescence-based, LIVE/DEAD (Life Technologies, Carlsbad, CA, USA) yeast viability assay as an indicator of the desired anti-apoptotic effect of the identified compounds. 18 Together, these assays lead to the identification of a single compound (from a small library of 2000) that inhibited apoptosis in the Vpr-expressing S. pombe.
Similar lethal expression approaches have been used in yeast-based screens for modulators of human cell processes.10–11 For example, expression of human p38α, a kinase with major implications in oncogenesis and inflammatory disease, proved toxic to S. cerevisiae. Quantification of yeast growth, again using optical density, was used in a phenotypic rescue HTS to screen a library of 40,000 compounds to identify 2 compounds that restored growth. Critically, both of these compounds inhibited activity in vitro and in mammalian-cell secondary assays, thereby validating the yeast-based primary assay as a screening approach. 20
Despite the validation of this approach for mammalian and viral targets, there are no reports of yeast lethal expression platforms being developed towards NTD drug discovery, although there are likely to be clear opportunities in this direction.
Transactivation
The HTS readout for lethal expression–based platforms relies on positive growth changes in response to the direct chemical modulation of the target protein. The unparalleled genetic tractability of the model yeast S. cerevisiae and S. pombe, however, allows the cell to be engineered to provide an indirect readout via transactivation. S. cerevisiae transactivation systems have been developed and used in HTS to identify inhibitors of human β-secretase, the enzyme that drives the production of Aβ peptides thought to be associated with Alzheimer’s disease, 21 and disruptors of the formation of a human calcium channel. 22 The transactivation approach is, however, best and most extensively illustrated by the work of the Hoffman Laboratory (Boston College) toward the discovery of mammalian phosphodiesterase (PDE) inhibitors. 23 Mammals express approximately 100 tissue-specific PDE isoforms that catalyze the conversion of cyclic adenosine monophosphate (cAMP) to 5′AMP and cyclic guanosine monophosphate (cGMP) to 5′GMP; consequently, regulating the levels of these cyclic secondary messengers influences a wide variety of cellular processes in a tissue-specific manner. Given these specific effects, the selective modulation of PDE enzymes has been recognized as having therapeutic potential for a wide range of human diseases.24–25
Unlike S. cerevisiae, S. pombe can tolerate deletion of adenylate cyclase (Csg1 in yeast). To create a yeast-based assay platform, csg2-null S. pombe were engineered such that expression of the selectable marker URA4 was placed under the control of the fbp1 promoter. This promoter was suppressed when the accumulation of cAMP or cGMP, caused by the loss of Csg1, activated protein kinase A. Under these conditions, the yeast are able to grow in the presence of 5-fluoroorotic acid (5FOA), which is converted to toxic fluoroorotidine monophosphate in the presence of URA4.23,26 Expression of either the yeast Csg1 or a mammalian PDE in this line, however, resulted in conversion of cAMP to 5′AMP or cGMP to 5′GMP, leading to URA4 expression and, consequently, 5FOA sensitivity. Inhibition of PDE or Csg1 rescued growth in the presence of 5FOA, allowing the yeast to be formatted into a 384-well, HTS-compatible, positive selection platform. This platform has been used to screen variously sized libraries (3000 to 200,000 compounds) for inhibitors of various human PDEs, with some hits demonstrating selective PDE inhibition and physiological activity (e.g., cAMP elevation) in secondary assays.26–28
Although there are no reports of transactivation technologies being used toward NTD drug discovery, clear opportunities exist. For example, protozoan PDE orthologues have been shown to be essential, and therefore putative drug targets, in several pathogenic species. 29 Formatting these into the S. pombe transactivation platform described above is clearly possible and could facilitate the search for novel inhibitors. The ability to counterscreen against human PDEs in the same platform would allow the identification of selective inhibitors that could represent lead compounds for NTD drug discovery.
Substitution
Although both lethal expression and transactivation platforms have been designed and used for HTS, the majority of yeast-based assays have relied on the simple substitution of an essential yeast function with an orthologue from the organism of interest. Key to this is the conservation of protein function between the yeast and an often distantly related eukaryote, such as mammals, helminthes, or protozoa.
The substitution approach is widespread in the screening of mammalian targets. 17 For example, a 10,000-compound library has been screened against a S. cerevisiae mutant complemented by the expression of a human potassium channel protein (Kir2.1). 30 By measuring liquid culture growth in 96-well plates, compounds inhibiting at low but not high potassium concentrations were selected, with those that inhibited growth at both concentrations discarded as nonspecific and generically cytotoxic. Such counterscreening is critical to eliminate off-target hits in cell-based assays. Furthermore, secondary assays demonstrated the selectivity of the identified inhibitors for Kir2.1 over other human potassium channels, with a single compound showing good activity in a mammalian neuronal cell line. 30 This multiscreening approach illustrates the role that relatively simple and inexpensive yeast-based HTS can play in primary HTS and counterscreening, identifying hits from a large library for more complex and resource-expensive secondary screening and validation.
In contrast to both lethal expression and transactivation platforms, the potential of yeast-based substitution platforms has been demonstrated for both helminth and protozoan drug discovery efforts. For example, with a view to discovering potential agents for the treatment of obesity, inhibitors of the human acetyl-CoA carboxylase 2 (AAC2) were identified from a 34,000-compound library using an 96-well HTS assay relying on S. cerevisiae in which the endogenous AAC coding region was replaced with the human AAC2 by homologous recombination. 31 To ensure selectivity for human AAC2, yeast complemented with human AAC1 as well as wheat and protozoan (Toxoplasma gondii) AACs were constructed. Notably, because AAC has been indicated to be a drug target in T. gondii 32 and the causative agent of the NTD human African trypanosomiasis, the protozoa Trypanosoma brucei, 33 this successful yeast-based AAC HTS 31 could be developed to identify selective inhibitors of the protozoal enzymes.
In addition to the validation of yeast substitution platforms that could be deployed for parasite (including NTD) drug discovery, a small number of papers report the direct development and use of these systems for HTS in this area.
Although not the cause of an NTD, the nematode worm Haemonchus contortus is a major parasite of ruminants and may be compared to the causative agents of the NTDs dracunculiasis (guinea-worm disease), lymphatic filariasis (elephantiasis), onchocerciasis (river blindness), and soil-transmitted helminthiases. 1 Ornithine decarboxylase (ODC) is a key regulatory enzyme in the biosynthesis of polyamines (PA) in eukaryotes and has been established as the protozoal target for difluoromethylorthine (DFMO; eflornithine), a central therapy for the NTD human African trypanosomiasis. 34 Using an S. cerevisiae ODC-deficient mutant complemented by expression of the H. contortus orthologue and formatted into a 96-well-plate HTS platform, a library of 90,000 compounds was screened for the ability to inhibit cell growth in the absence of PA. 35 Significantly, and in contrast to the assays described above, Alamar Blue was used as an indicator of yeast cell growth, which enabled the output to be measured colormetrically. Alamar Blue, however, also produces a fluorescent signal corresponding to cell growth, which can provide a more sensitive output than that obtained from optical assays. To eliminate nonselective compounds, hits from this primary screen were then counter-screened in the presence of PA, conditions in which ODC is redundant. 35 The single compound identified failed, however, to inhibit the helminth ODC in vitro instead, perhaps inhibiting a yeast enzyme involved in spermidine and spermine synthesis. 35 This emphasized the importance of secondary screening and the potential functional redundancy in yeast that could confound the interpretation of yeast-based HTS data.
Until recently, this report by Klein et al. 35 was the only published work reporting the use of yeast-based HTS in the search for novel antiparasitics. As discussed above, though, the utility of this type of platform has been demonstrated for the protozoan drug target AAC. 31 Building on previous work in which S. cerevisiae was used as a tool to study the Plasmodium falciparum drug target dihydrofolate reductase (DHFR), 36 however, mutant yeast have been complemented by the expression of DHFR from protozoan and helminth causes of NTDs. 37 In this agar plate-based assay, the antimalarial DHFR inhibitor pyrimethamine inhibited the growth of yeast complemented with DHFR from drug-sensitive but not drug-resistant P. falciparum, thus confirming target specificity. In the same system, the DHFRs from the NTD causative agents Leishmania major (protozoa; leishmaniasis), T. brucei (protozoa; human African trypanosomiasis), T. cruzi (protozoa; Chagas disease), and Schistosoma mansoni (helminth; schistosomiasis) proved less sensitive to pyrimethamine. 37 To develop this as a platform for HTS, Bilsland et al. engineered mutant yeast to express fluorescently tagged, complementing DHFR protozoan orthologues, as well as other mutant lines complemented by similarly tagged versions of the drug targets N-myristoyltransferase (NMT) and phosphoglycerate kinase (PGK). 38 Judicious use of fluorescent tags facilitated the multiplexing of the assay, allowing the protozoan DHFR, NMT, or PGK from each species to be screened simultaneously with the human orthologue, in a single well of a 384-well plate, against each member of the Maybridge Hitfinder Library (14,400 chemically diverse compounds), thus identifying parasite-specific hits. Of the 36 specific hits identified in this assay, 18 proved cytotoxic to T. brucei in cell culture. 38
This study by Bilsland et al. 38 currently represents the only published report of a yeast-based HTS being used for NTD drug discovery, and it should be noted that the multiplexed counterscreen against yeast complemented with the human orthologue should help ensure that the activity of the identified compounds is target specific, although confirmatory secondary biochemical assays are also required.
Using a similar substitution strategy, the protozoan drug target inositol phosphorylceramide (IPC) synthase has been characterized.39–41 IPC synthase, which catalyzes the formation of a nonmammalian phosphosphingolipid, is a 6 transmembrane domain protein and as such is refractory to conventional biochemical analyses. Given this, partially purified microsomal extracts from complemented yeast have been used to develop a 96-well plate-based enzyme that was used to define the mode of action and substrate requirements of the protozoan enzyme.42–43 This assay platform is, however, unsuitable for true HTS due to its complexity, requiring ion exchange to separate fluorescently tagged lipid substrate from fluorescently tagged lipid product. To facilitate high-content HTS, IPC synthase (AUR1p) 44 null S. cerevisiae complemented with the L. major orthologue has been formatted into a robust 1536-well assay platform and used to screen the GSK compound collection, identifying selective submicromolar inhibitors of the parasite enzyme 45 (https://www.openlabfoundation.org). 47 Signifi-cantly, to the best of our knowledge, the IPC synthase screen represents the largest yeast-based screen to date, for both NTDs and other disease targets.
Potential Problems and Solutions
As outlined above, the use of yeast as a vehicle for drug discovery has several advantages compared to in vitro target-based and conventional cell-based approaches. These include the ability to easily manipulate the platform for various counterscreening approaches and the low cost and simplicity of growth, coupled with the ability to screen a target within a cellular context. The system has certain problems that need to be considered, however, when designing a yeast-based HTS and interpreting the data output (Table 1).
First, and importantly, although yeast provides a eukaryotic context for the target, this context remains axenic and may not reflect the in vivo scenario. In addition, when using a substitution platform (the only examples described for parasite targets), the target protein must obviously have a functional yeast orthologue. However, the future development of lethal expression and transactivation systems for antiparasitic discovery may overcome this barrier.
A second issue, as identified above in the screen for helminth ODC inhibitors, 35 is the possibility of functional redundancy in yeast. Although HTS and counterscreening against mutant yeast complemented with the parasite ODC identified one apparently selective compound, this failed to inhibit the helminth ODC in vitro, an effect that indicates it inhibited an alternative yeast pathway for PA biosynthesis. 35 This demonstrated that the issue of functional redundancy must be considered in relatively simple eukaryotes as well as in complex systems, 10 and it emphasized the necessity to validate identified hits in secondary (biochemical) assays. The genetic tractability of yeast has, however, facilitated the multiplexing of an HTS substitution platform to simultaneously screen a compound library against parasite targets and their human orthologues. 38 The ability to discriminate between a target and an orthologue under exactly the same assay conditions in a straightforward manner allowed the rapid selection of target-selective hits, although secondary assays (biochemical and cellular) remain desirable to validate the compounds.
Last, a further impediment to the use of yeast as a vehicle for HTS is the presence of extremely effective drug efflux systems. 10 Yet again, however, the tractability of yeast has facilitated some sophisticated solutions. In S. cerevisiae, deletion of the efflux pump Snq2 enhanced sensitivity of a human phosphoinositide kinase-3 lethal expression assay, 46 and ablation of the major multidrug export pump encoded by PDR5 similarly increased the sensitivity of a T. brucei DHFR-complemented mutant yeast to pyrimethamine. 37
Discussion
As eukaryotic cell-based but target-directed platforms for HTS, yeast platforms offer several advantages over both in vitro target-based and conventional cell-based assays (Table 1); and, as discussed above, the tractability of yeast facilitates genetic-engineering solutions to most of the perceived problems. Furthermore, the assays are robust, as determined by a Z’-factor greater than 0.5.18,23,46 These facts, coupled with the relative simplicity and low cost of yeast-based HTS, indicate that this technology would be ideally applied to drug discovery for parasitic NTDs. Currently, efforts in this domain are limited, although the multiplexed format designed and used for screens against drug targets from several protozoan and helminth NTDs may provide the impetus to drive forward future work. 38 In this respect, it is important to note that these assays have been widely miniaturized into 384-well plates18,23,38 and more recently into a 1536-well format for uHTS (https://www.openlabfoundation.org). 47 In addition, the multiplexing of assays for inhibitors of antiparasitic targets with the human orthologues has facilitated the rapid identification of target-specific hits, 38 although, as evidenced above, 35 in all cases hits from a primary yeast HTS should be validated using subsequent counterscreens and secondary assays to confirm the molecular mode of action. 31
The hit rate in yeast-based primary assays appears highly variable among targets, platforms, and diversity libraries. For example, the 0.03% of 1.8 million compounds seen for the Leishmania inositol phosphorylceramide synthase assay (https://www.openlabfoundation.org) 47 was comparable to that seen with a similar substitution platform and the mammalian AAC (0.02% of 30,000 compounds). 31 In contrast, screening against the Leishmania and Trypanosoma DHFR in a substitution platform gave a hit rate of 0.25% (of 14,400 compounds), 38 similar to that seen for mammalian PDE assayed using a transactivation platform (0.3% of 200,000 compounds). 27 The reasons, beyond target and library choices, for this dramatic variation are unclear, although differences in the sensitivity of response detection, optical density 31 versus fluorescent protein markers, 38 and the selection cutoff will clearly influence the hit rate. This variation in assay and hit rate is sustained in secondary enzymatic or cell-based assays; for example, 18 of the 36 hits in the primary assay for protozoan DHFR were cytotoxic to T. brucei in cell culture (an overall hit rate of 0.13% of 14,400 compounds), 38 compared with 4 of 595 initially selected compounds proving to be selective PDE11 inhibitors, with only one demonstrating the expected cellular function (an overall hit rate of 0.0005% of 200,000 compounds). 31 The second case, however, used much more complex and comprehensive secondary and tertiary screening protocols, whereas the first provided no direct evidence of DHFR inhibition.
In summary, the potential utility of yeast to serve as a vehicle for protozoan and helminth NTD drug discovery is clear, and an idealized yeast-based HTS protocol is illustrated in Figure 1. This flexibility ensures that a wide variety of targets, including those regarded as less tractable (e.g., transmembrane proteins), can be screened in uHTS in a robust and cost-effective manner.
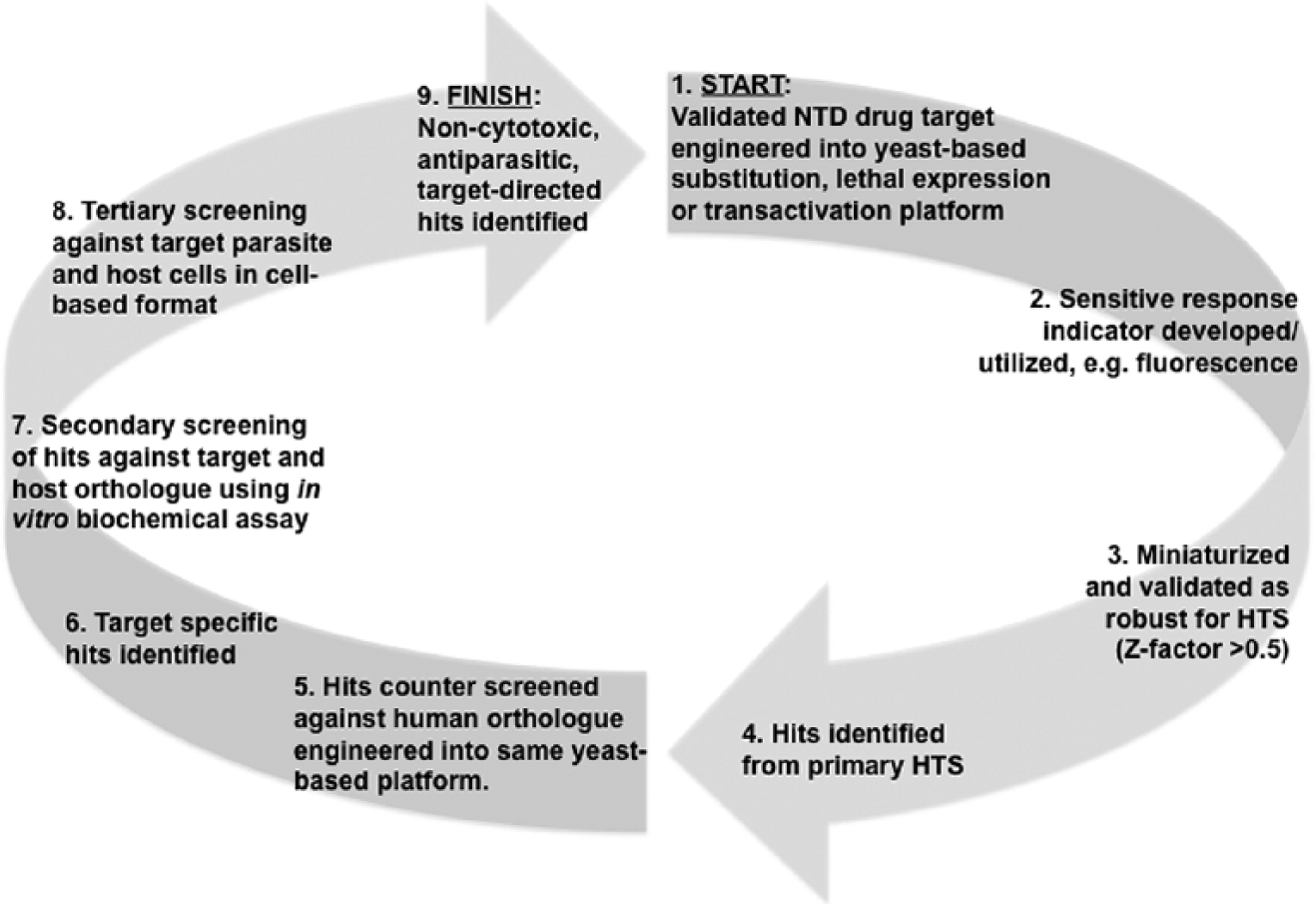
An idealized scheme for the use of a yeast-based platform for early-stage, anti-NTD (neglected tropical disease) hit discovery via high-throughput screening. Numbered 1 (START) to 2 (FINISH). Beyond target validation and the primary assay (1–4), it is key to use counterscreening (5) to identify target-specific hits (6), which can then be validated as such in in vitro biochemical assays (7). Subsequently, these compounds must be validated as antiparasitic and noncytotoxic in cell-based assays (8–9).
Footnotes
Acknowledgements
We would like to acknowledge the support of the Open Lab Foundation (PWD and PGS).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We would like to acknowledge the support of the Open Lab Foundation (PWD and PGS).