
Abstract
In this article, the authors describe a colorimetric, high-throughput assay suitable for optimizing the activity of the recently discovered sulfotransferase LipB, by directed evolution. Crucially, LipB uses para-nitrophenol sulfate as donor in the sulfation of the nucleoside antibiotic liposidomycin B-I and other acceptor surrogates. Thus, using a robotic liquid-handling device, crude cell extracts were prepared from an Escherichia coli strain that overproduced LipB in wells of a microplate, and production of para-nitrophenol at 405 nm was monitored spectrophotometrically. Enzyme activity could be detected only in the presence of both LipB substrates and overexpressed LipB. The screen displays a suitable standard deviation for directed evolution and importantly is not limited to the natural desulfo-liposidomycin acceptor. The authors plan to use the screen to identify LipB variants with altered acceptor specificity and promiscuity for use in sulfation of natural products and other small-molecule therapeutics.
Keywords
Introduction
E
Critical to the success of directed evolution is the design and implementation of a suitable high-throughput screen (HTS) or selection to identify those rare variants that are improved in some way. 10 With regard to modification of natural products, for example, the development of a simple HTS based on glycosylation of a fluorescent surrogate acceptor substrate 11 yielded mutant glycosyltransferases with incredible promiscuity for the glycodiversification of small-molecule therapuetics. 12 In another example, an agar-based diffusion assay was used to improve the activity of the acyltransferase LovD for the semi-synthesis of simvastitin by directed evolution. 13 In addition, a well-established HTS for cytochrome P450s was harnessed to identify variants for the regioselective deprotection of monosaccharide substrates. 14 Yet, HTSs or selections are not available for many enzymes that modify or synthesize natural products, and this limits the scope of evolutionary methods for overcoming stringent substrate specificity.
One group of enzymes that modify natural products for which HTSs for directed evolution are not available is the sulfotransferases (STases). Exemplary sulfated natural products include micafungin, a clinically approved antifungal drug, the unusual high solubility of which is ascribed to sulfation. 15 Other prominent classes of sulfated natural products include the chlorosulfolipids, 16 antibacterial sulfated glycopeptides, 17 protein tyrosine kinase inhibitors such as the sulfated steroids halistanol, 18 trypsin inhibitors such as the cyclic depsipeptide micropeptin 90, 19 potent antifungals such as the sulfated polyhydroxypolyene amphidinol, 20 antifouling sulfated phenolic acids, 21 sulfated terpenes with various antimicrobial activities,22,23 and sulfated hydroquinones that inhibit HIV reverse transcriptase. 24
In addition, the fatty acyl nucleoside antibiotics comprise several members that are sulfated, including the A-90289s 25 and the liposidomycins ( Fig. 1 ). Related members of this family, such as caprazamycin, are potent inhibitors of bacterial translocase I, 26 and represent exciting targets for the development of new antibiotics to overcome resistance to current antibiotics. 27 Recently, the sulfotransferase responsible for the installation of sulfate into liposidomycin B-I from the producing organism Streptomyces sp. SANK 60405 was identified by gene deletion and characterized in vitro, revealing the unusual preference for p-nitrophenol sulfate (pNPS) as the sulfate donor ( Fig. 2 ). 28 Moreover, it was found that LipB could also use the readily available uridine as sulfate acceptor, in place of the presumed desulfo-natural acceptor. 28 Although STases usually use 3′-phosphoadenosine-5′-phosphosulfate as a sulfate donor, the discovery that LipB uses pNPS raises the unique possibility of using the production of the well-known chromophore p-nitrophenol (pNP) to screen the activity of a natural product STase ( Fig. 2 ). Here, we report the validation of An HTS for the directed evolution of LipB. The motivations for potentially engineering the specificity of LipB by directed evolution are twofold. First, to produce new liposidomycin analogues by fermentation and precursor directed biosynthesis, it is likely that the promiscuity of LipB needs to be enhanced because sulfation is used as a self-resistance strategy that leads to the in vivo inactivation of liposidomycins as well as that of the A-90289s. Second, it may be possible to employ evolutionary methods to further improve the specificity of LipB to more distantly related acceptors, to produce libraries of sulfated small molecules for drug discovery, particularly if An HTS could eventually be adapted to an ultra high-throughput format.
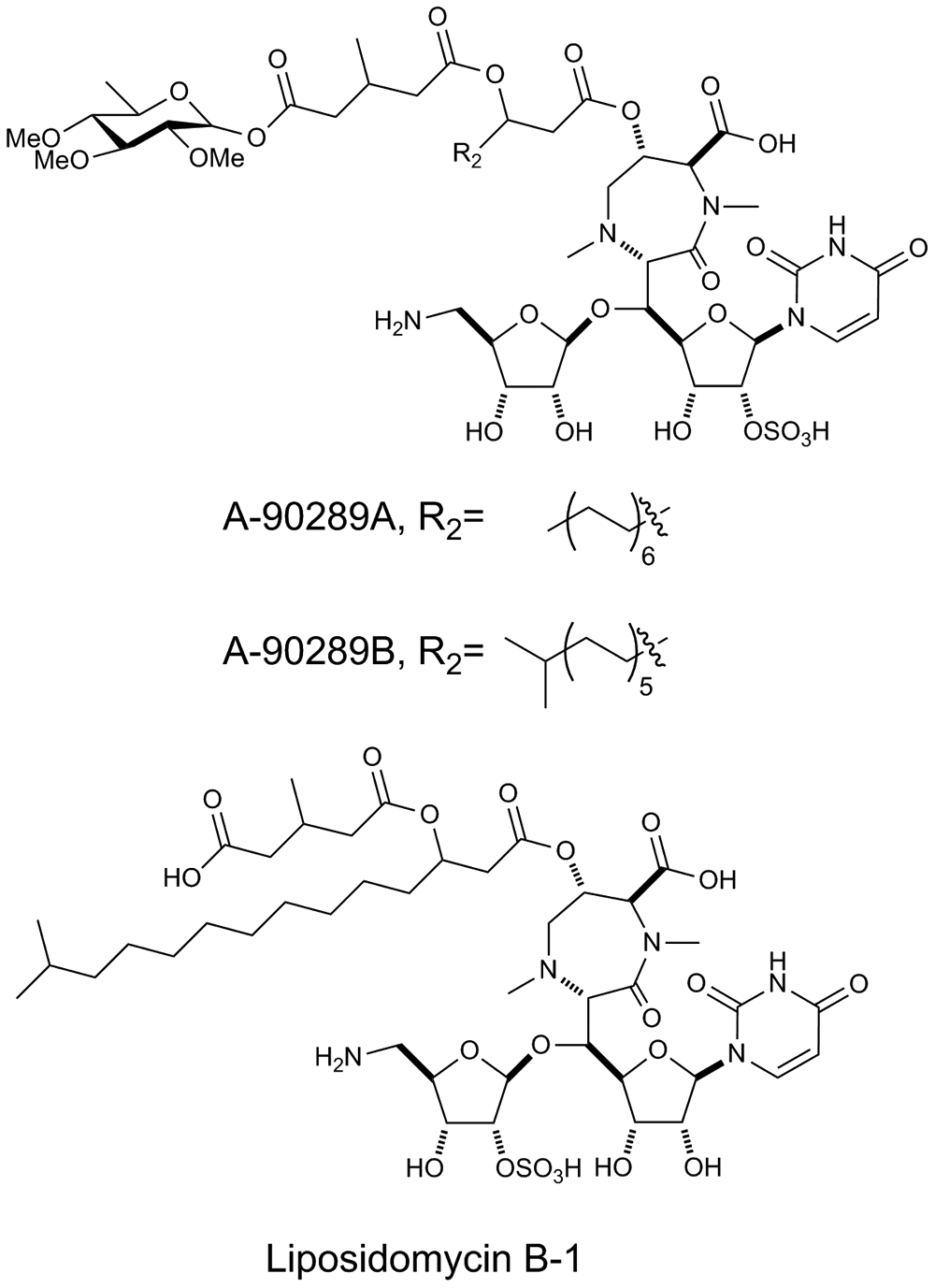
Examples of sulfated nucleoside antibiotics. Although the published structure of liposidomycin B-1 indicates sulfation at the 2″ position, this is in fact more likely to occur at the 2′ position. 28
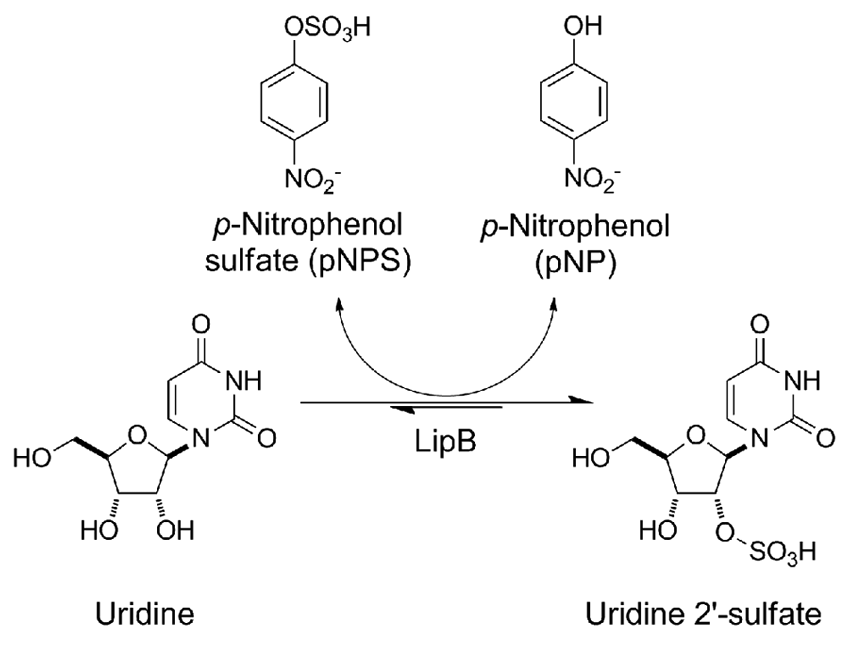
Proposed high-throughput screen for reporting activity of the STase LipB. LipB catalyzes the sulfation of uridine (a surrogate for the presumed desulfo-liposidomycin acceptor), with the production of p-nitrophenol (pNP), which can be monitored spectrophotometrically at 405 nm.
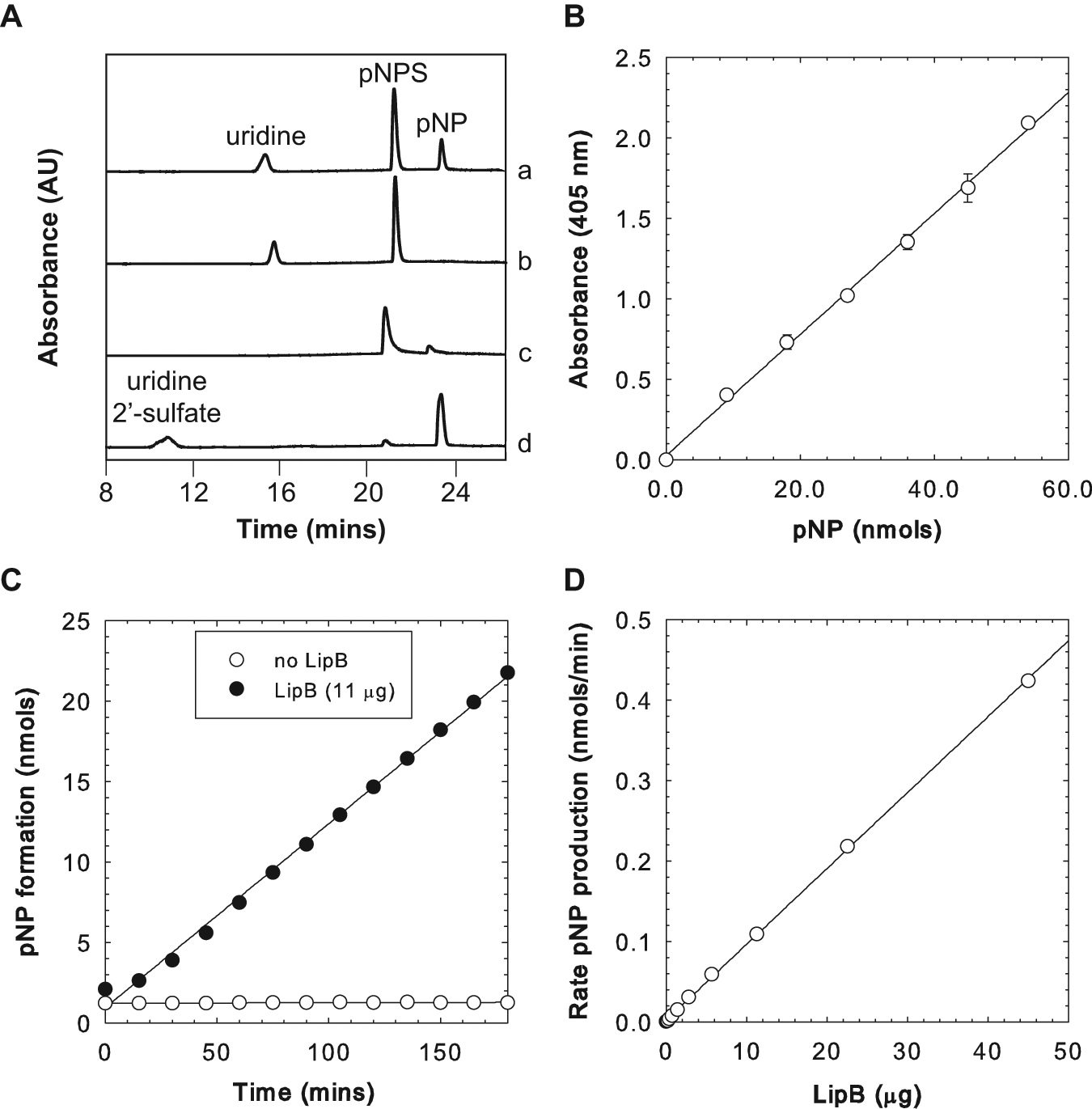
(
Materials and Methods
Unless otherwise stated, all materials and reagents were of the highest grade possible and purchased from Sigma (St. Louis, MO). IPTG was from Calbiochem (Gibbstown, NJ). Bacterial strain Escherichia coli BL21(DE3) pLysS competent cells was from Promega (Madison, WI). Primers were ordered from Integrated DNA Technologies (Coralville, IA). Analytical high-performance liquid chromatography (HPLC) was performed on a Varian ProStar system (Palo Alto, CA). Mass spectra were obtained using electrospray ionization on a Thermo TSQ Quantum Discovery MAX connected to a UV/Vis diode array detector. For LC-MS analysis, quenched reaction mixtures were analyzed by analytical reverse-phase HPLC with a 150 × 0.5 mm Zorbax SB-C18 5 µm column (Agilent, Santa Clara, CA) using a gradient of 2% acetonitrile in 0.2% formic acid/H2O and 98% acetonitrile in 0.2% formic acid/H2O at 15 µL/min, with detection at 254 nm (see below for HPLC gradient).
Cloning of LipB into pET28a
The gene for LipB from Streptomyces sp. SANK 60405 was amplified from pET30-LipB 28 using the oligonucleotides LipB-for (5′-GCGTGCCATATGGTCCGGACGAGAAC-3′) and LipB-rev (5′-AGC CGCAAGCTTTCAGCGCACCCGCGC-3′) and ligated into pET28a via the NdeI and HindIII restriction sites (underlined). The DNA sequence was confirmed by sequencing. The deposited lipB gene sequence contains an erroneous N-terminal extension (MTV), corresponding to an incorrectly located start codon. The corrected sequence was deposited at NCBI (Accession no. BAJ05878.2).
Mutant library construction
The random mutant library was prepared via error-prone PCR using the Stratagene GeneMorph II Random Mutagenesis Kit, as described by the manufacturer, using 1 µg of pET28a-LipB as template. The primers used for amplification of the lipB gene were LipB-for and LipB-rev. Amplified product was digested with NdeI and HindIII, purified by agarose gel electrophoresis (0.8% w/v agarose), extracted using the QIAquick Gel Extraction Kit (QIAgen, Valencia, CA), and ligated into similarly treated pET28a. The ligation mixtures were transformed into chemically competent NovaBlue cells and single colonies used to prepare plasmid for DNA sequencing, which revealed that the library had the desired mutation rate of 3.5 nucleotide mutations per gene. In addition, restriction digestion analysis of randomly selected clones indicated that 90% of the library members contained gene insert. Subsequently, all the transformants from this library were pooled and cultured overnight. Plasmid was prepared from this culture and used to transform chemical competent E. coli BL21(DE3)pLysS, which was screened as described below.
Expression and purification of LipB protein
E. coli BL21(DE3) pLysS competent cells were transformed with the plasmid pET28a-LipB, and positive transformants were selected on LB agar supplemented with 30 µg/mL kanamycin. A single colony was transferred to 3 mL LB supplemented with 30 µg/mL kanamycin and grown at 37 °C and 250 rpm for 10 h. Then, a 0.5 mL aliquot was transferred to 50 mL LB supplemented with 30 µg/mL kanamycin and incubated at 37 °C and 250 rpm overnight, then 5 mL of the culture was used to inoculate 500 mL LB supplemented with 30 µg/mL kanamycin. The 500 mL culture was incubated at 18 °C and 250 rpm and protein expression induced at OD600 ˜0.6 by the addition of isopropyl β-D-thiogalactoside (IPTG) to a final concentration of 0.1 mM. After incubation at 18 °C and 250 rpm for 14 h, cells were collected by centrifugation at 5000g for 20 min and resuspended in 20 mL of 100 mM Tris-HCl pH 8.0 containing 300 mM NaCl and then lysed by sonication. Following centrifugation at 10 000g, the soluble extract was loaded onto a 1-mL HisTrap HP column (GE Healthcare, Piscataway, NJ) and purified by fast protein liquid chromatography using the following buffers: wash buffer (20 mM phosphate [pH 7.4] containing 0.5 M NaCl and 20 mM imidazole) and elution buffer (20 mM phosphate [pH 7.4] containing 0.5 M NaCl and 500 mM imidazole). The purified protein was concentrated using an Amicon Ultra 30 000 MWCO centrifugal filter (Millipore, Billerica, MA) and stored as 10% glycerol stocks at −80 °C. Protein purity was verified by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Protein quantification was carried out using the Bradford Protein Assay Kit from Bio-Rad (Hercules, CA).
HPLC assay
In vitro enzyme assays were performed in a 300-µL reaction mixture containing 50 mM sodium phosphate (pH 8), uridine (1.5 mM), p-nitrophenylsulfate (2 mM), and LipB (9 mg/mL) at 25 °C. Aliquots were removed and quenched with an equal volume of ice-cold methanol and centrifuged at 10 000 g for 10 min, and cleared supernatants were used for HPLC analysis. A series of linear gradients was developed from 0.1% TFA (A) in water to methanol (HPLC grade, B) using the following protocol: 0 to 8 min, 100% A; 8 to 18 min, 60% B; 18 to 25 min, 95% B; 25 to 32 min, 95% B; 32 to 35 min, 100% A. The flow rate was 1 mL/min, and the absorbance was monitored at 260 nm using Pursuit XRs C18 column (250 × 4.6 mm; Varian). The expected sulfated uridine was characterized by LC-MS, C9H12N2O9S calculated 323.03 [M-H]−, observed 323.8.
Expression of LipB protein in a 96-deep-well microplate
An Eppendorf epMotion liquid-handling machine (Hauppauge, NY) was used for liquid transfer steps. Individual colonies of BL21(DE3)pLysS pET28a-LipB or BL21(DE3)pLysS pET28a were used to inoculate wells of a round-bottomed 96-deep-well plate (VWR) containing 1 mL LB medium supplemented with 30 µg/mL kanamycin. Culture plates were tightly sealed with AeraSeal breathable film (Research Products International Corp., Prospect, IL) and incubated at 37 °C and 350 rpm for 18 h. Of each culture, 100 µL was transformed to a freshly prepared deep-well plate containing 1 mL of LB medium supplemented with 30 µg/mL kanamycin. The freshly inoculated plate was incubated at 37 °C and 350 rpm for 3 h, at which point protein expression was induced with 1 mM IPTG. The plate was incubated for 18 h at 20 °C and 350 rpm. Cells were harvested by centrifugation at 5000g for 10 min and resuspended in 250 µL of 100 mM Tris-HCl pH 8.0 buffer containing 300 mM NaCl and 10 mg/mL of lysozyme. The plates were then subjected to a single cycle of freeze/thaw, and the cell debris were collected by centrifugation at 5000g for 10 min; 50 µL of each cleared extract was used for the reaction.
Colorimetric assay
For the HTS reaction, 50 µL of cleared extract was added to 250 µL 50 mM sodium phosphate buffer (pH 8) containing uridine (1.8 mM) and p-nitrophenylsulfate (2.4 mM). Upon mixing, the absorbance at 405 nm was measured using a BioTek Hybrid Synergy 4 plate reader (Winooski, VT) and incubated for up to 6 h, and the absorbance measurements were repeated. Alternatively, assays were also performed using various amounts of pure LipB.
Results
We aimed to extend well-known enzyme assays based on pNP production 29 for use in screening large libraries of LipB variants. For directed enzyme evolution, enzyme variants are usually expressed from E. coli within wells of a microplate and crude cell extracts prepared in situ for enzyme assay. As a first step toward this goal, we set out to replicate the activity of purified LipB, which would be free of any potentially contaminating enzyme activities that might otherwise contribute to or reduce LipB activity in crude extracts. Accordingly, we subcloned the gene for LipB from pET30-lipB into pET28a, which would be a more convenient vector for subsequent random mutagenesis (see the Materials and Methods section). The protein was purified to homogeneity by metal affinity chromatography (data not shown), and the enzyme activity was determined by low-throughput HPLC. Incubation of purified LipB with both pNPS and the surrogate acceptor uridine resulted in the production of a new product peak, as detected by HPLC ( Fig. 3A ). Product identity was confirmed by mass spectrometry (see the Materials and Methods section). We noted that a low level (<5% the pNP production rate with uridine) of pNPS hydrolysis was observed when uridine was omitted from the reaction mixture, consistent with LipB-catalyzed hydrolysis in the absence of acceptor. With confirmation that the activity of LipB could be reconstituted in vitro, we next established the linear range of detection of pNP in a microplate by spectrophotometry. As anticipated, pNP could be easily detected at >10 nmol under our assay conditions, and the absorbance response was linear within this range ( Fig. 3B ). Next, the activity of purified LipB was determined by following the absorbance at 405 nm over time. Gratifyingly, the production of pNP could easily be detected above background levels measured in the absence of enzyme ( Fig. 3C ). Repeating this assay with various amounts of LipB established that the rate of pNP production was linearly dependent on the total LipB content when up to 45 µg protein was used ( Fig. 3C ). We estimated that aliquots of crude cell extract used for our directed evolution experiments would likely contain <10 µg LipB, as judged by SDS-PAGE (data not shown). We note that at the highest amount of LipB tested (45 µg), the rate of pNP production corresponds to <1% total conversion of uridine to sulfated product, which in turn would correspond to a kcat of <1 min−1 (assuming saturation). Although low, this likely accurately reflects the level of activity expected for enzymes from secondary metabolism, especially considering the use of a surrogate acceptor substrate (uridine) in our assay.
Next, we turned our attention to validating the HTS using crude cell extracts. Accordingly, extracts were prepared from small-scale cultures of E. coli BL21(DE3) that harbored either pET28a-lipB or pET28a, as positive and negative controls, respectively, using lysozyme and freeze-thaw treatment (see the Materials and Methods section). Subsequent colorimetric assay for pNP production revealed that activity of LipB could be easily distinguished from the negative control lacking overexpressed LipB in crude extracts ( Fig. 4A ). In addition, activity was completely dependent on the presence of both uridine and pNPS, as assays that lacked either or both substrates showed levels of activity that could not be distinguished from that with extracts prepared using the negative control.
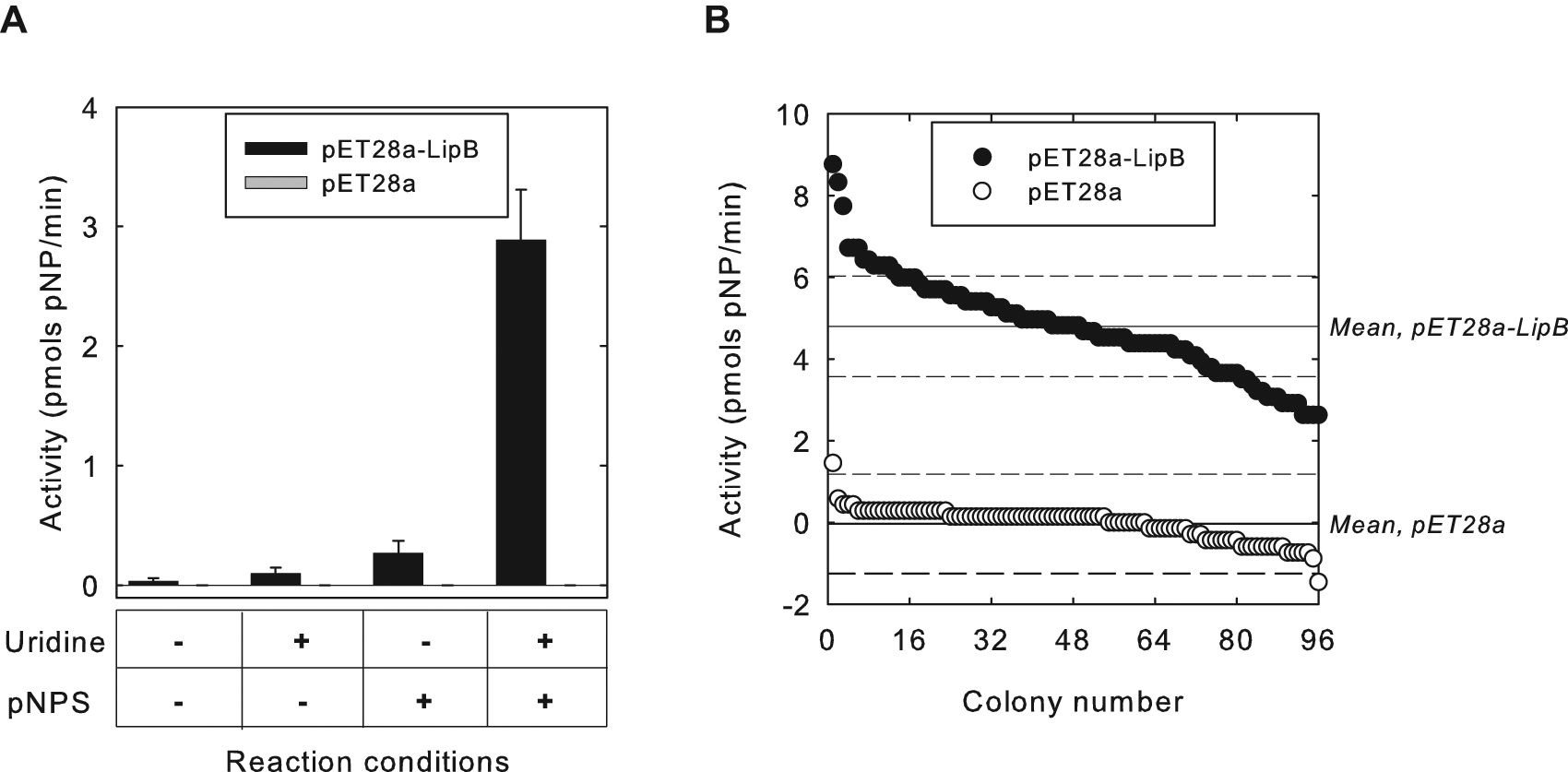
(
To further establish that this assay was reliable for HTS in microplate format, we next assayed the activity of a total of 96 samples each of positive and negative controls by inoculating colonies of the respective E. coli strain into individual wells of a microplate. We used a robotic liquid-handling device to carry out all the liquid transfer steps of the cell culturing, crude extract preparation, and assay setup. Colorimetric assay for pNP product resulted in a standard deviation of ˜20% for the extracts containing LipB ( Fig. 4B ), perfectly adequate for directed evolution experiments. Indeed, every positive well produced a higher level of activity than any negative control well, even provided the activity of LipB in crude cell extracts is rather low. Cumulatively, these data demonstrate that LipB activity can be faithfully detected in crude extracts prepared in microplates by our colorimetric assay.
The HTS was then used to characterize the activities of a small number (200 colonies) of LipB variants created by error-prone PCR. The random mutagenesis conditions were adjusted to achieve a mutagenesis rate of 3.5 nucleotides per gene, corresponding to one to two amino acid mutations per gene product. The activities of ˜200 library members are shown in Figure 5 , displayed in descending order. In contrast to the data presented for the control strains ( Fig. 4A , B ), the library contained mutants with a broad range of activities, and as might be expected, several library members (˜50%) did not display any detectable activity with uridine and pNPS, as defined in Figure 4B . Although a portion of the library displayed equal or higher activity compared with wild-type (WT) LipB, variants with significantly higher activity (e.g., greater than 2 standard deviations higher than the WT activity) were not identified.
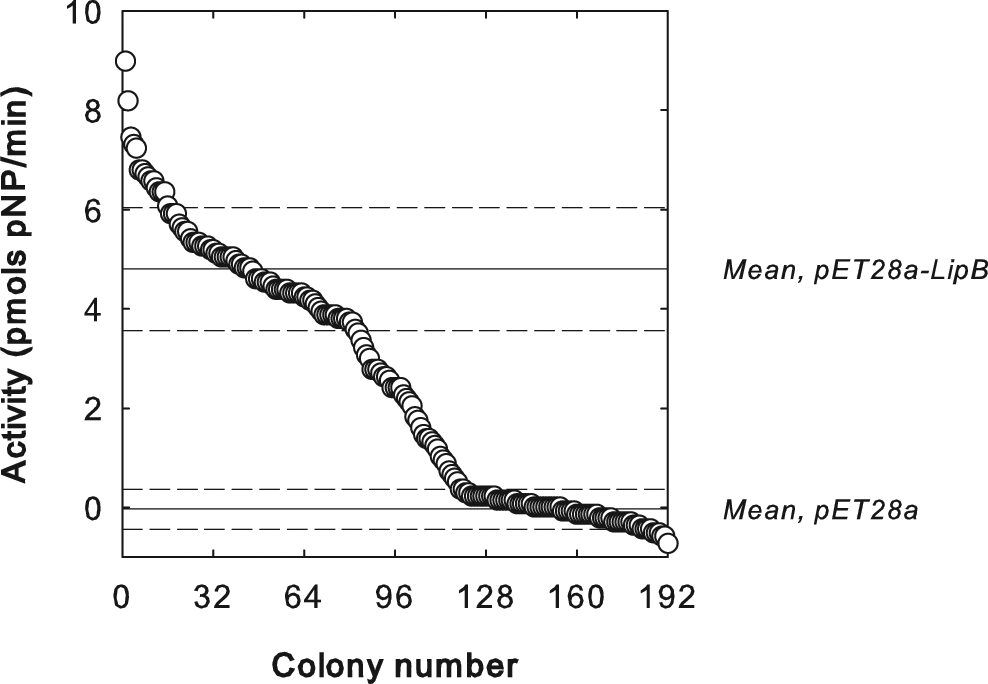
Activities of clones from a library of LipB variants produced by error-prone PCR, plotted in descending order. Solid horizontal lines indicate mean activity for pET28a-LipB and pET28a clones; dashed horizontal lines indicate 1 standard deviation ± the mean.
Discussion
Although assays for determining the activity of STases involved in detoxification of xenobiotics have been described, 30 to the best of our knowledge, this study reports for the first time an HTS adapted for use toward directed evolution of a STase, whereby crude bacterial extracts provide the mutant enzyme. Crucially for directed evolution, this HTS is easy to perform, and several thousand colonies can be processed in a week. Furthermore, the ability of LipB to use pNPS as a sulfate donor makes this assay system unique with respect to the prospect of creating promiscuous STases for the modification of natural products by directed evolution.
The HTS described here allows screening with a broad range of substrates not limited to uridine or the presumed natural sulfate acceptor desulfo-liposidomycin. This feature is particularly important given our motivations for developing the HTS. We carried out a number of assays using both purified LipB and enzyme expressed in crude cell extracts to demonstrate the validity of the proposed HTS. In addition, we also demonstrated the utility of our HTS by screening the activity of ˜200 mutant LipBs created by error-prone PCR. Mutant LipBs with significantly improved activity were not identified in this small library. Indeed, screening several thousand mutants is usually required to identify such improvements.12,13
Given the rather low level of LipB activity that we observed in crude extracts, our first target for directed evolution will involve improving the overall activity of LipB with uridine and pNPS as substrates, prior to altering the specificity toward alternative nonnatural substrates. This goal may be achieved by improving protein expression, enzyme solubility, or catalytic activity via random mutagenesis and screening. In the long term, we aim to use both whole-gene random mutagenesis and semi-random mutagenesis, guided by sequence information and homology models, in combination with our HTS to identify LipB mutants with altered specificity or promiscuity.