
Abstract
Posttranslational modifications of histone tails are very important for epigenetic gene regulation. The lysine-specific demethylase LSD1 (KDM1A/AOF2) demethylates in vitro predominantly mono- and dimethylated lysine 4 on histone 3 (H3K4) and is a promising target for drug discovery. We report a heterogeneous antibody-based assay, using dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA) for the detection of LSD1 activity. We used a biotinylated histone 3 peptide (amino acids 1–21) with monomethylated lysine 4 (H3K4me) as the substrate for the detection of LSD1 activity with antibody-mediated quantitation of the demethylated product. We have successfully used the assay to measure the potency of reference inhibitors. The advantage of the heterogeneous format is shown with cumarin-based LSD1 inhibitor candidates that we have identified using virtual screening. They had shown good potency in an established LSD1 screening assay. The new heterogeneous assay identified them as false positives, which was verified using mass spectrometry.
Introduction
The study of inherited changes in phenotype or gene expression without any changes in the underlying DNA sequence is called epigenetics. Major epigenetic changes are cytosine methylation in DNA and posttranslational modification of histones. These modifications have an essential role in transcription regulation and lead to gene silencing or activation depending on the modification and the context. Methyl marks on lysines are installed by lysine methyl transferases (KMTs), which can result in mono-, di-, or trimethylated states. As counterplayers, there are also demethylases that are divided into two classes according to their mechanism of demethylation. One class is composed of the iron and α-ketoglutarate–dependent jumonji-C domain-containing lysine demethylases (JmjC), and the other are the FAD-dependent lysine-specific demethylases 1 and 2 (LSD1, LSD2). 1 While JmjC domain-containing enzymes can also accept trimethylated lysines as substrates, LSD1 can demethylate only mono- and dimethylated H3K4 2 and, in a complex with other epigenetic enzymes and the nuclear androgen receptor (AR), H3K9. 3
In AR-responsive tumor cells, demethylation of H3K9 by LSD1 leads to transcriptional activation of AR-dependent genes.
3
This suggests that LSD1 is a target for the treatment of androgen-dependent tumors such as prostate cancer. Since LSD1 expression is also strongly increased in other cancers,
1
LSD1 has become a prominent and promising target in cancer drug discovery. The first inhibitors of LSD1 were identified by their structure and sequence similarity to monoamine oxidases A and B (MAO) that are well-studied antidepressant and anti-Parkinson drug targets. One of these inhibitors is (±)-trans-2-phenylcyclopropan-1-amine hydrochloride (
Available high-throughput screening (HTS) assays for LSD1 are homogeneous with fluorescence readout and thus vulnerable to artifacts caused by intrinsic fluorescence or quenching by the library compounds. One principle is the detection of hydrogen peroxide, which is generated by the FAD-dependent oxidation of the substrate. The hydrogen peroxide is in turn a cosubstrate in a second reaction with horseradish peroxidase (HRP) to form a fluorophore, which can then be detected. We used Amplex Red (Invitrogen, CA) as a substrate for HRP, which is converted to resorufin and can be detected with excitation/emission maxima at 570/585 nm or 530/590 nm. An alternative is the detection of formaldehyde that is generated by the oxidative demethylation process. Formaldehyde is oxidized by formaldehyde dehydrogenase to formic acid, thereby converting NAD+ to NADH, which can be detected with excitation/emission maxima of around 330/460 nm.8,9 For these assays, usually a H3K4(me2) or H3K4(me1) substrate with the first 21 amino acids of histone 3 is used, but full-length histone H3 works as well. 8
Other commercially available homogeneous assays (e.g., based on AlphaLISA or LANCE [PerkinElmer, Waltham, MA]) use a biotinylated H3K4(me1) (aa1–21) substrate. The detection of the level of methylation is achieved by specific antibodies against unmodified H3K4 (H3K4(me0)), followed by a time-resolved fluorescence readout. As weak inhibitors may also lead to quenching in these proximity-based systems, we were looking for a heterogeneous LSD1 assay, primarily as a follow-up screen for screening hits from one of the different homogeneous formats.
Here we present the development of a new heterogeneous system based on a dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA) to detect LSD1 activity and inhibition in vitro. Due to its heterogeneous nature, the assay is not vulnerable to fluorescence or quenching by library compounds. DELFIA assays have the advantage of time-resolved fluorescence detection with a large Stokes shift (difference between excitation and emission wavelengths) and high sensitivity, resulting in a very good signal-to-noise ratio. We demonstrate that our assay can be used to test reference inhibitors and potential new LSD1 inhibitors that were identified from a virtual screening campaign. The new assay showed that the identified coumarins were false positives.
Materials and Methods
(±)-trans-2-Phenylcyclopropan-1-amine hydrochloride (PCPA, tranylcypromine) and HRP were purchased from Sigma Aldrich (Steinheim, Germany); namoline was from InterBioScreen Ltd. (Moscow, Russia); 96-well DELFIA streptavidin-coated yellow plates (AAAND-0005), DELFIA enhancement solution (1244-105), and DELFIA europium-labeled anti–mouse antibody (AD0124) were from PerkinElmer; biotinylated H3K4(me1) and H3K4(me2) (both aa1-21-GGK-biot) were from Peptide Specialty Laboratories Ltd. (Heidelberg, Germany); and anti-H3K4(me0) monoclonal mouse antibody (05-1341, lot NG1844755) was from Millipore (Billerica, MA). Time-resolved fluorescence was measured on an EnVision (PerkinElmer), with settings adopted from the standard PerkinElmer DELFIA protocol. Pipetting was performed on a PerkinElmer Janus automated workstation or manually. Washing steps were undertaken with a Tecan (Crailsheim, Germany) hydroflex microplate washer or manually, each 6 times with 1:10 diluted 10× TRIS wash buffer, pH 7.5 (here DELFIA wash buffer, from Solid Phase Guide, second edition [2001], Nunc Brand Products, Dreieich, Germany). Antibody incubation was performed in a TRIS incubation buffer (TRIS wash buffer + 0.5% bovine serum albumin [BSA]). LSD1 enzyme was expressed in Sf9 cells as published elsewhere. 3 Amplex Red was obtained from Invitrogen.
Fluorescence Spectra
In total, 20 µL of the 10-mM compound stock solution was dissolved in 1 mL demethylation buffer (containing 45 mM HEPES, 40 mM NaCl, pH 8.5). The spectra were acquired on a PerkinElmer LS 45 with a fixed excitation wavelength at 365 nm. The spectra range was set from 380 to 620 nm. As a blank control, the demethylation buffer (containing 2% DMSO) was used.
HRP Coupled Assay
Kinetic measurement was performed with 12 µL LSD1, 20 µL Amplex Red mixture containing 100 µM Amplex Red reagent, and 2 U/mL HRP in demethylation buffer (containing 45 mM HEPES, 40 mM NaCl, pH 8.5), DMSO (2% final), and 5.8 µL demethylase buffer in a 384-well white opaque microplate. With 20 µM biotinylated H3K4(me1) peptide, the reaction was started and measured on a BMG (Offenburg, Germany) POLARstar microplate reader (λex: 550 nm, λem: 615 nm) for 50 min.
IC50 values were determined with 6 µL LSD1 (respectively an amount that leads to 60%–80% intensity of the fluorescence intensity measured from a 10-µM solution of resorufin), 2 µL inhibitor in DMSO/demethylase buffer (2% final DMSO), 20 µM H3K4(me2) peptide, and demethylation buffer in a total volume of 20 µL for 15 min. Then, 20 µL of Amplex Red mixture was added and immediately measured.
DELFIA Assay
In total, 6 µL LSD1 (respectively an amount that leads to 60%–80% intensity of the fluorescence intensity measured from a 10-µM solution of resorufin), 4 µL demethylase buffer, and 2 µL inhibitor or blank solution (containing 20% DMSO and 80% buffer v/v, 2% DMSO final) were added to an Eppendorf tube and shaken for 5 min at room temperature (RT). Peptide solution (2.2 µL peptide [20 µM/well] plus 5.8 µL demethylation buffer) was added to start the reaction. After a 15-min incubation at 30 °C, the reaction was stopped by adding 380 µL DELFIA wash buffer (pH 10) and shaken to give a homogeneous solution. Then, 5 µL of this solution and 95 µL DELFIA wash buffer (pH 10) were added to a 96-well streptavidin-coated yellow microplate. After incubation for 1 h with shaking, the wells were washed six times with DELFIA wash buffer. Next, 100 µL of a dilution of anti-H3K4(unmodified) antibody in TRIS incubation buffer (1:10,000) was added and again incubated for 1 h at 30 °C. After the same washing procedure as before, incubation with 100 µL europium-labeled anti–mouse antibody in TRIS incubation buffer (1:250) at 30 °C for 1 h was started. After a last washing step, 100 µL enhancement solution was added and, after 10 min of shaking at RT, measured in a PerkinElmer EnVision with the recommended filters and mirrors from PerkinElmer. For different batches of each antibody, it might be necessary to adjust dilutions to obtain a maximum signal window. For the determination of the Z′ factor, we used 15 positive controls (enzyme, substrate, and DMSO, no inhibitor, see above; maximum signal) and 15 negative controls (instead of DMSO, 200 µM of PCPA in DMSO; minimum signal).
Virtual Screening
Due to the structural similarity between LSD1 and the monoaminoxidase B (MAO-B), we used the known MAO-B inhibitor cumarin-2 (7-[(3-chlorobenzyl)oxy]-2-oxo-2H-chromene-4-carbaldehyde), which also had been co-crystallized with MAO-B (PDB code 2V60 10 ), as a starting point. In the crystal structure, cumarin-2 makes electrostatic and Van der Waals interactions with Tyr326, Leu171, Gln206, Ile199, and Leu164. Hydrogen bonds are formed between the oxygen belonging to the aldehyde moiety and the side chain of Tyr435. Cumarin-2 was used for a similarity screening using the Chembridge compound collection (590,029 compounds) and MACCS fingerprints within MOE2011.10. A Tanimoto coefficient of 0.85 was taken as a cutoff value. Twenty-five hits were retrieved and docked into the MAO-B (2V60.pdb) and LSD1 (2DW4.pdb) binding pocket. For docking, GOLD 4.1 software (Cambridge, UK) and GoldScore were used. Top-ranked compounds that showed the same interaction (calculated with the MOE protein-ligand interaction fingerprint) as cumarin-2 were retrieved and purchased.
Mass Spectrometry
The demethylation assays were performed essentially as described. 2 In total, 1 µg of peptide corresponding to the H3 tail residues 1 to 20 carrying two methyl groups at K4 was incubated with 0.3 to 5 µg of bacterially expressed and purified His-LSD1. The crude demethylase reactions were diluted 1:10 in a saturated α-cyano-4-hydroxycinnamic acid solution (50% acetonitrile, 0.1% trifluoroacetic acid) and spotted onto a matrix-assisted laser desorption ionization (MALDI) target plate. For each reaction, 2000 spectra were recorded and analyzed using the Data Explorer Software (Matrix Science, London, UK).
Results and Discussion
A major problem in antibody-based assays, especially for the quantitation of lysine methylation, is an antibody with good selectivity. We therefore immobilized unmodified, monomethylated, and dimethylated H3K4 (aa1-21-GGK-biot) to streptavidin-coated yellow microtiter plates (1 h, 37 °C, TRIS incubation buffer, 100 µL, 200 nM peptide) and measured the selectivity of different anti-H3K4(me2) antibodies (data not shown). Since all of the tested antibodies were generated in rabbits, a europium-labeled secondary anti–rabbit antibody was used to quantify the amount of bound first antibody (1 h, 37 °C, TRIS incubation buffer, 1:500 diluted). Although several of the used anti-H3K4(me2) antibodies (Millipore H07-030, lot DAM1724042; Active Motif [Carlsbad, CA] 39141, lot 01008001; Cell Signaling [Danvers, MA] 9725) from previous lots 11 had been shown to possess good selectivity over monomethyl lysine, in our hands, they were not selective over H3K4(me1), but the antibodies from Millipore and Active Motif showed selectivity over H3K4(me0), with a better sensitivity and signal-to-noise ratio for the Active Motif antibody. The current lot of the Cell Signaling antibody failed to show any selectivity against the different modifications in our hands. We then tested a monoclonal anti-H3K4(unmodified) mouse antibody (Millipore, 05-1341, lot NG1844755) against H3K4(me0), H3K4(me1), and H3K4(me2) and again detected binding with a DELFIA europium-labeled anti–mouse secondary antibody. The antibody showed good selectivity and signal-to-noise ratio ( Fig. 1A ). Since we could now detect the direct formation of the product H3K4(me0) and LSD1 can demethylate H3K4(me1) as well as H3K4(me2), we decided to set up the assay procedure with this antibody and the monomethylated H3K4(me1, aa1-21-GGK-biot) as the substrate.
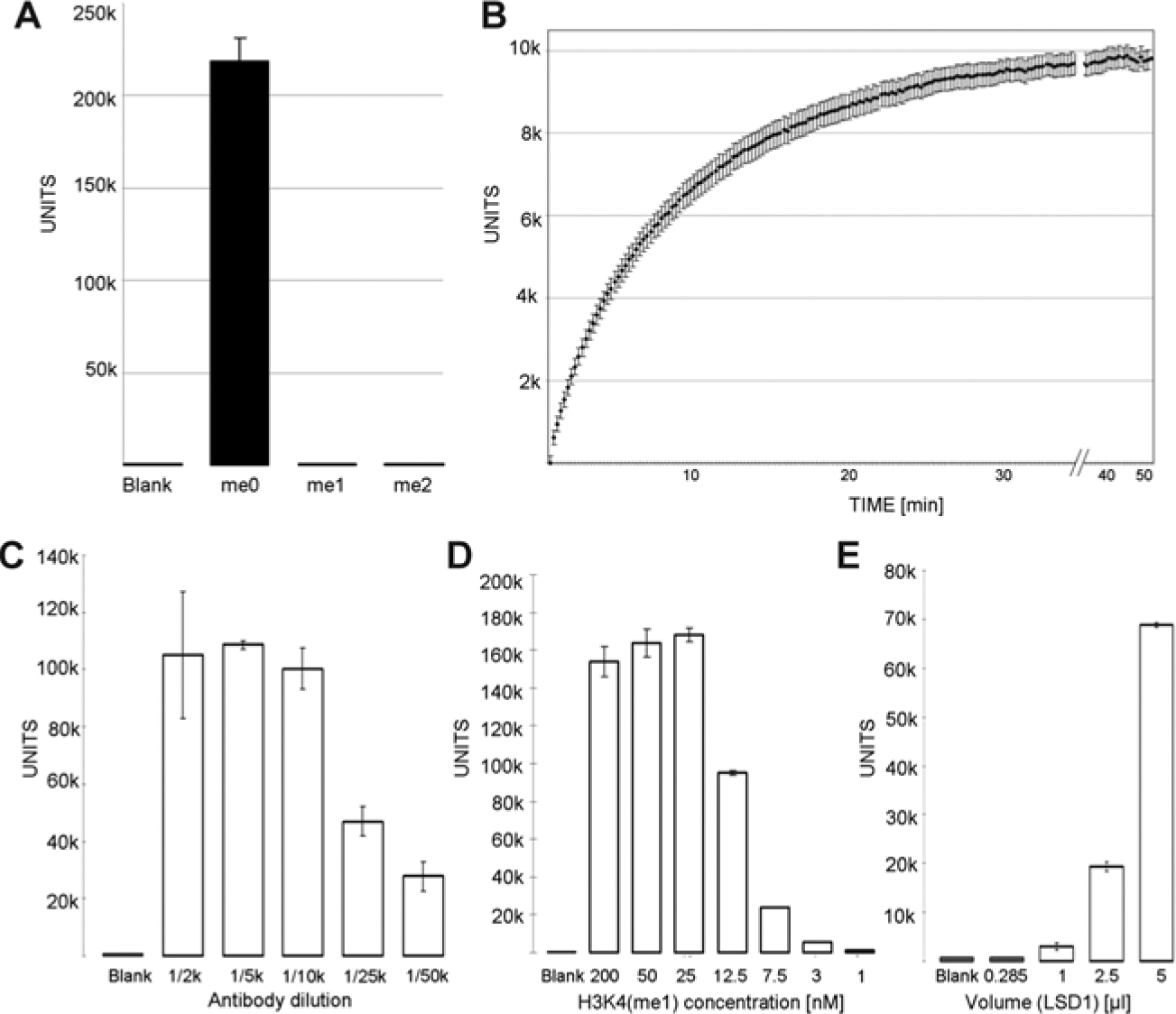
(
We immobilized the H3K4(me1-biot) substrate to streptavidin-coated 96-well microplates, but no demethylation by LSD1 could be observed. Since commercially available homogeneous assays (AlphaLISA and LANCE) use the same substrate for their assays, probably the proximity to the surface of the wells prevented demethylation by LSD1. We ensured conversion of the biotinylated H3K4(me1) substrate in an established Amplex Red assay (data not shown). As the experiment confirmed biotinylated H3K4(me1) to be an LSD1 substrate in solution, incubation before binding to the streptavidin-coated plates was then employed for the heterogeneous assay design.
We then optimized the concentrations of the primary and secondary antibodies. We first titrated the primary antibody with constant amounts of secondary antibody (1:250) and peptide (200 nM, 100 µL). We observed that even at primary antibody dilutions of 1:10,000, we still monitored a sufficient signal ( Fig. 1C ). To further ensure that primary antibody binding was saturated, the peptide was titrated down to 1 nM and the decrease in signal was measured ( Fig. 1D ). We observed a decrease in signal intensity between 25 and 12.5 nM, indicating that 12.5 to 25 nM H3K4(me0) peptide can be quantified with this amount of primary antibody.
We then tested different enzyme concentrations of LSD1 in the assay. The assay was performed as described in the Materials and Methods, but with increasing amounts of LSD1 ( Fig. 1E ). A sufficient signal could be seen after 20 min when 2.5 µL LSD1 was applied and an even better one with 5 µL of LSD1 preparation. With optimized conditions, we performed an HRP-coupled assay to test kinetics ( Fig. 1B ). From these results, we decided to stop the enzyme reaction after 15 min in the DELFIA assay.
To validate this assay, we tested PCPA (
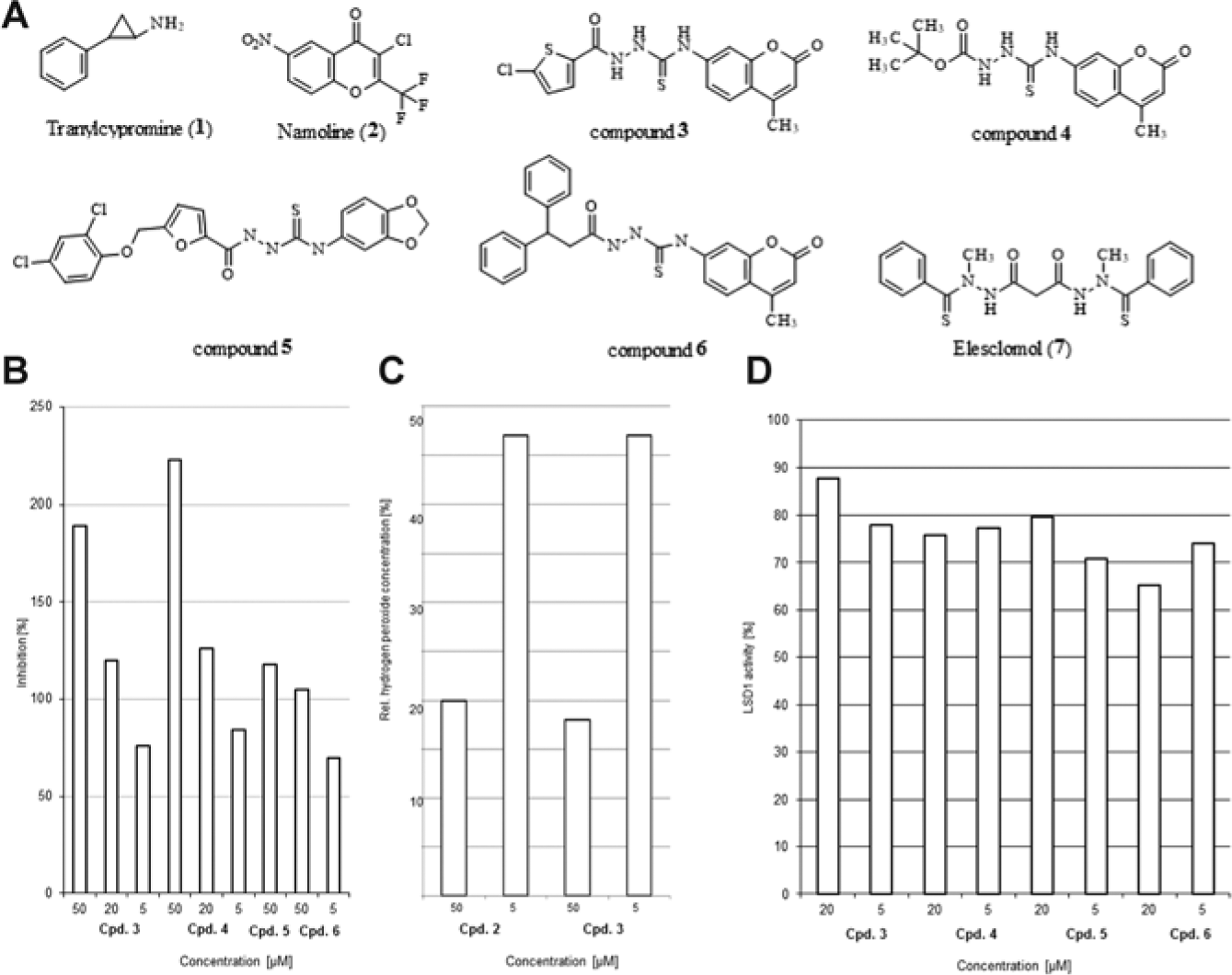
(
To analyze the influence of biotinylation on the demethylation reaction by LSD1, we tested PCPA in an HRP system using the biotinylated H3K4(me1) substrate. The IC50 value changed from 16.2 ± 2.3 µM with unbiotinylated H3K4(me2) peptide to 4.18 ± 0.20 µM, suggesting that the biotinylated substrate leads to decreased IC50 values but the values are observed in a similar range of potency. We also determined a Z′ factor for the assay, which is 0.7 and thus considered excellent (see
We then applied our new heterogeneous assay to some potential new LSD1 inhibitors that we had shortlisted for testing by virtual screening. As namoline had been identified as an LSD1 inhibitor based on a protein similarity analysis followed by focused library screening of an MAO-based chromone library, we applied a similarity-based approach to look for a new LSD1 inhibitor among cumarins as established MAO inhibitors. They generally cannot be investigated in the formaldehyde dehydrogenase assay due to intrinsic fluorescence (see
The analysis of the available X-ray structure of MAO-B co-crystallized with a known cumarin derivative (cumarin-2, PDB 2V60
10
) and the docking poses obtained for the virtual screening hits
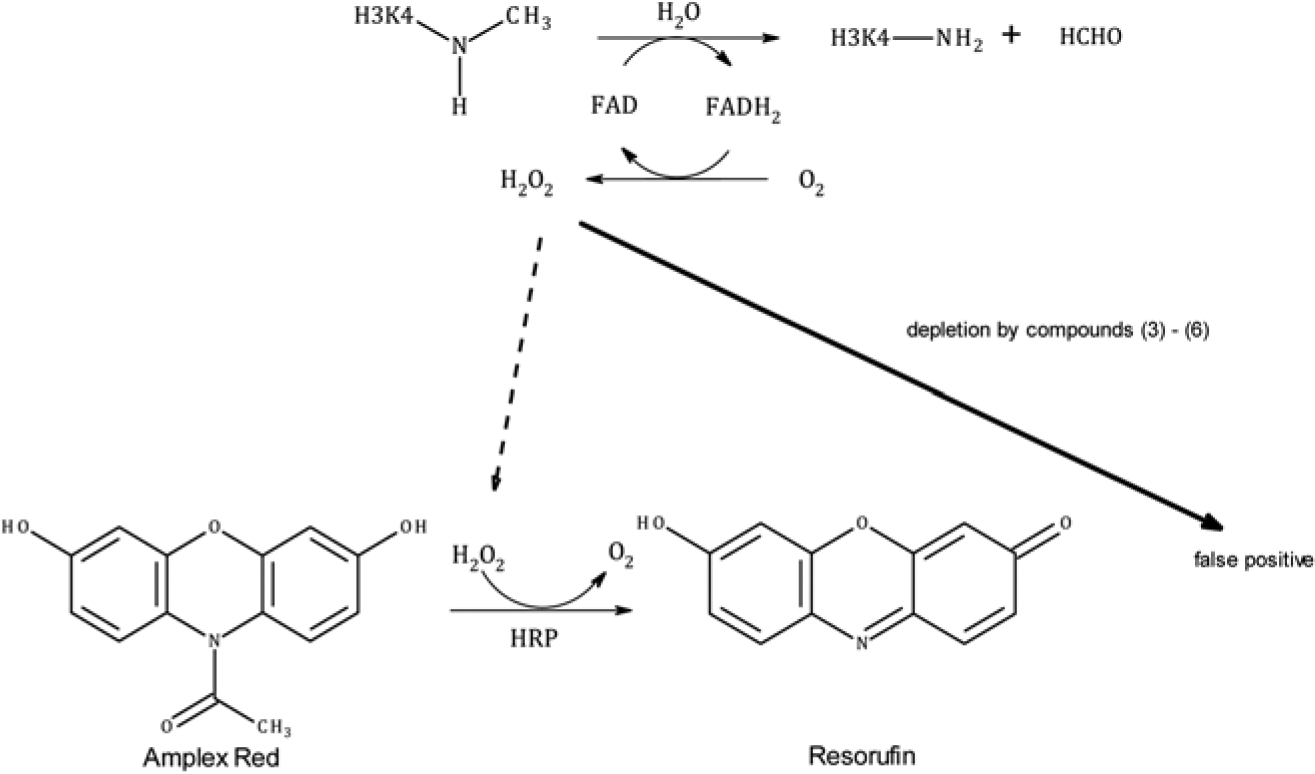
The upper panel shows the reaction of the LSD1-catalyzed demethylation of monomethylated lysine 4 from an H3 histone peptide. The reaction leads to the formation of H2O2, which is detected by oxidation of Amplex Red to resorufin. If library compounds deplete H2O2 by chemical reaction, no formation of resorufin occurs, which is also the case if LSD1 is inhibited. Thus, reactive compounds mimic LSD1 inhibition as false positives that can be unmasked using our new heterogeneous assay design.
These results were in contrast to the prediction from the virtual screening. Due to the larger binding cavity of LSD1 (1700 Å) compared with MAO-B (637 Å), it is possible that the candidate inhibitors adopt further interaction modes that are not able to block the enzymatic activity.
14
Due to the limitations of current scoring functions, it might be possible that the selected docking poses are not preferred ones. Another reason might be the higher polarity of the VS hits compared with the known MAO inhibitor cumarin-2 (logP 4.26 for cumarin-2, logP 2.83 for VS hit
In summary, our newly developed heterogeneous DELFIA assay is able to detect H3K4(me1) demethylation by LSD1 in vitro. It is a time-resolved fluorescence-based test system that is not vulnerable to assay interactions like compound fluorescence, quenching, or H2O2 consumption that interfere with other fluorescence-based LSD1 assays in homogeneous systems, like peroxidase or formaldehyde dehydrogenase-coupled detection principles. In addition, this assay design may be adaptable to other histone-modifying enzymes (e.g., Set7/9, an enzyme that is responsible for monomethylation of H3K4). 1
Footnotes
Acknowledgements
We thank A. Mai, University La Sapienza, Rome, for a gift of the reference LSD1 inhibitor MC2850. 15
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding by the Deutsche Forschungsgemeinschaft (assay development and virtual screening: DFG, M.J.: Ju295/7-1; W.S.: Si868/4-1; R.S.: Schu688/11-1) and the BMWi (cumarin based inhibitor testing: M.J. and R.S.; KF2389502AJ9) is gratefully acknowledged.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.