
Abstract
Acquiring sufficient quantities of iron to support survival is often a critical limitation for pathogenic bacteria. To meet this demand, bacteria have evolved unique strategies to scavenge iron and circumvent the nutritional immunity exerted by their hosts. One common strategy, which is often a key virulence factor for bacterial pathogens, involves the synthesis, secretion, and reuptake of iron chelators known as siderophores. In vitro and in vivo studies have demonstrated that the siderophore aerobactin is critical for virulence in the hypervirulent pathotype of Klebsiella pneumoniae (hvKP). Given the high rate of multidrug resistance in K. pneumoniae, and in light of the ever-increasing demand for novel Gram-negative therapeutic targets, we identified aerobactin production as a promising antivirulence target in hvKP. Herein, we describe the development of a high-throughput biochemical assay for identifying inhibitors of the aerobactin synthetase IucA. The assay was employed to screen ~110,000 compounds across several commercially available small-molecule libraries. IucA inhibitors with activity at micromolar concentrations were identified in our screening campaigns and confirmed using secondary orthogonal assays. However, the most potent compounds also exhibited some properties commonly observed with promiscuous/nonspecific inhibitors, including incubation time and target enzyme concentration dependence, as well as the potential to antagonize unrelated enzymes.
Keywords
Introduction
Iron is a critical nutrient that plays a variety of structural and functional roles in biological systems. It is involved in redox chemistry in multiple enzyme systems, sometimes as a lone cofactor, as in the iron- and α-ketoglutarate-dependent oxygenases, or in the context of larger molecular complexes, such as those found in heme-dependent enzymes and iron-sulfur clusters. Due to the low solubility of iron oxidized to the ferric state, bioavailable iron is limiting in many physiologic environments encountered by microorganisms. Microbes have therefore evolved to produce a wide array of siderophores, small-molecule iron chelators that are secreted into the environment where they form high-affinity chelation complexes with ferric ions. 1 These iron–siderophore complexes are then assimilated back into the cell through specialized uptake systems, whose expression is often co-regulated with the siderophore biosynthetic genes.
A limited number of conserved iron-chelating functional groups are incorporated into a variety of structurally diverse siderophores. Iron chelation is most commonly mediated by the oxygen atoms of carboxylate, catechol/phenol, and hydroxamate moieties that decorate different types of scaffolding. Many bacteria use nonribosomal peptide synthetases (NRPSs) to produce siderophores. NRPSs use a fascinating modular enzyme architecture to produce peptides without the use of mRNA or ribosomes. 2 Further, the ability of the NRPSs to utilize nonproteinogenic amino acids and aryl acids enables the incorporation of these iron-binding chemical functionalities. Common NRPS-derived siderophores include enterobactin, which is present in many enteric Gram-negative species; pyoverdine and pyochelin from Pseudomonas; mycobactin from mycobacteria; and bacillibactin from Bacillus species. 1
The NRPS-independent siderophores (NISs) constitute a second major class of siderophores. 3 These compounds are most commonly composed of hydroxamate constituents linked together by polyanionic citrate, α-ketoglutarate, or succinate molecules. A shared feature of the NIS biosynthetic pathways is the presence of ATP-dependent ligases that couple the hydroxamate-bearing building block to a carboxylate of the di-/tri-acid backbone. 4 As with NRPS siderophores, NISs are found in both Gram-positive bacteria, including staphyloferrin of Staphylococcus and desferrioxime of Streptomyces, and Gram-negative bacteria, including achromobactin of Pectobacterium, and aerobactin, which is produced by a number of Enterobacteriaceae species. Interestingly, some Bacillus species produce petrobactin, a hybrid NRPS-NIS stealth siderophore. 1
The necessity of micromolar concentrations of iron to support microbial growth makes siderophore production an important phenotype of many bacteria. Targeting siderophore systems has therefore been examined from many angles for potential antivirulence treatment approaches, including obstructing ferric siderophore import systems, 5 blocking siderophore uptake and cycling with rigidified analogues and mimetics, 6 neutralization via siderophore-based immunization strategies,7,8 increasing drug potency and specificity through siderophore-antibiotic “Trojan horse” conjugates, 9 and antagonizing siderophore biosynthetic enzymes. 10 Multiple studies support the strategy of targeting siderophore systems by demonstrating that genetic or chemical disruption can result in the attenuation of microbial growth and virulence under iron-limiting conditions.10,11 We have previously used a high-throughput biochemical screening approach to identify inhibitors of pyoverdine production by blocking the fatty acid hydrolase critical for maturation of the pyoverdine chromophore. Inhibitors with Ki values in the submicromolar range were capable of blocking pyoverdine production in iron-depleted culture conditions. 12
While Klebsiella pneumoniae has long been recognized as a common nosocomial pathogen, hypervirulent strains of K. pneumoniae (hvKP), which have the ability to infect otherwise healthy people in the community, are receiving renewed attention by the medical community. 13 These hvKP strains were initially described in Southeast Asia, but have now been reported all around the globe. Phylogenetic analysis revealed that nearly all hvKP isolates harbor a ~224 kB plasmid containing multiple virulence-related genes. 14 Of significant concern, a contemporary report from China described the transfer of this virulence-conferring plasmid to a carbapenem-resistant K. pneumoniae strain. 15 The coalescence of drug-resistant and hypervirulent phenotypes offers the possibility of an untreatable and highly virulent pathogen.
Analysis of the genetic markers of hvKP that contribute significantly to its virulence revealed that iron acquisition plays a major role.16,17 Most hvKP strains are capable of producing four siderophores, the NRPS siderophores yersiniabactin and enterobactin, the glycosylated enterobactin derivative salmochelin, and the NIS aerobactin. Genetic studies demonstrate that mutant hvKP strains unable to produce aerobactin showed significantly reduced virulence in subcutaneous and intraperitoneal mouse infection models compared with either the parental wild-type organism or strains mutated in any or all of the other siderophore biosynthetic pathways.16,17 The critical role that aerobactin plays in virulence has prompted our effort to understand its biosynthesis and develop tools that could further validate aerobactin production and/or utilization as a novel antivirulence target.
The aerobactin operon encodes four biosynthetic enzymes (IucABCD) and a transmembrane transporter (IutA) involved in aerobactin uptake.
18
IucD and IucB hydroxylate and acetylate
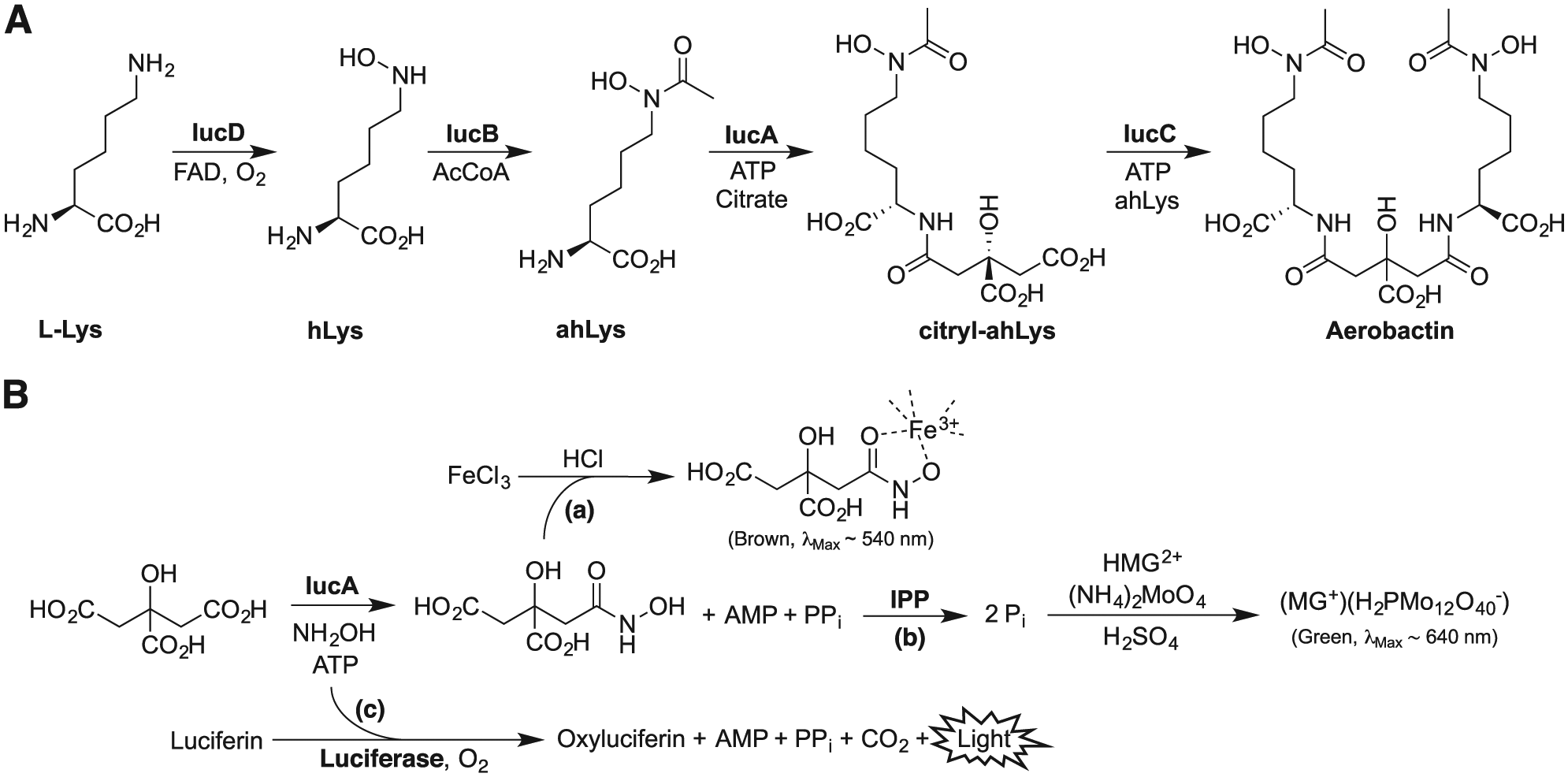
(
While there are multiple potential avenues to block the aerobactin system, we sought to leverage our recent structural and functional studies of IucA and target this critical enzyme for inhibition. A similar strategy was adopted to identify natural product inhibitors of the related NIS synthetases SbnE and AsbB of staphyloferrin B and petrobactin biosynthesis, respectively, identifying the baulamycins as micromolar inhibitors of the two target enzymes. 21 Bioactivity assays against both Gram-positive and Gram-negative species harboring NIS pathways were promising, showing IC50 values that ranged from 20 to 150 µM. However, the baulamycins were surprisingly equally effective at inhibiting growth in both iron-rich and iron-limiting conditions, suggesting that an off-target effect may predominate under the conditions investigated. Indeed, the general toxicity of the baulamycins was recently reported to stem from damage to bacterial membranes. 22 Toward the goal identifying a small-molecule probe capable of inhibiting IucA, we describe our high-throughput primary assay development, initial screening studies, and the secondary orthogonal assays used to evaluate the activity of inhibitors identified in our screening. Our studies identified several compounds that were active in the primary and secondary orthogonal assays at micromolar concentrations. However, initial follow-up analysis of the most potent compounds revealed some in vitro characteristics consistent with nonspecific enzyme interference mechanisms.
Materials and Experimental Methods
Materials
All commercial reagents were used as provided unless otherwise indicated. Inorganic pyrophosphatase (IPP; Saccharomyces cerevisiae), myokinase (MK; rabbit muscle), pyruvate kinase/lactate dehydrogenase enzymes (PK/LDH; rabbit muscle), adenosine triphosphate disodium salt hydrate (ATP), sodium citrate tribasic dihydrate, hydroxylamine hydrochloride, malachite green (MG) oxalate, ammonium molybdate, Tween 20, sulfuric acid, ferric chloride hexahydrate, acetohydroxamic acid, anhydrous DMSO, and SYPRO Orange were purchased from Sigma-Aldrich (St. Louis, MO). TCEP was purchased from Hampton Research, Inc. (Aliso Viejo, CA). The Kinase-Glo Plus assay kit was purchased from Promega Corp. (Madison, WI) and used according to the manufacturer’s instructions. Multiple commercial compound collections were screened for inhibitors of IucA. All libraries were sourced as 10 mM solutions in DMSO. An initial pilot screen of 4400 compounds was conducted using bioactive compounds from the Library of Pharmacologically Active Compounds (LOPAC; Sigma-Aldrich), Spectrum Collection (MicroSource Discovery Systems, Inc., Gaylordsville, CT), and Tocriscreen Total (Tocris Bioscience, Bristol, UK) chemical libraries. Larger-scale screening campaigns included the EXPRESS-Pick library (ChemBridge [CB] Corp., San Diego, CA) and the HitDiscover collection (Maybridge [MB], Thermo Fisher Scientific, Waltham, MA). Potential leads identified from the larger screening campaigns were reordered as solids directly from CB and MB, dissolved in DMSO to yield 10–20 mM stock solutions, and used without any further analysis or purification.
IucA Expression and Purification
The target enzyme in this study was aerobactin synthetase IucA from hypervirulent K. pneumoniae hvKP1 (GenBank EMB09144.1). The iucA gene was amplified from hvKP1 genomic DNA using primers to incorporate restriction sites at the 5′ and 3′ ends of the gene. The gene was subcloned into a modified pET15b vector containing an N-terminal 5xHis tag and a TEV protease recognition site. The expression vector was transformed into E. coli BL21(DE3) for protein production. Cells were grown in LB media at 37 °C (250 rpm) for approximately 3 h to an OD600 of ~0.65. IucA expression was induced with the addition of 500 µM IPTG, followed by incubation at 16 °C (250 rpm) for ~18 h. Cells were harvested by centrifugation at 6 × 103g for 15 min at 4 °C. After decanting the supernatant media, the cell pellet was flash frozen in liquid N2 and stored at –80 °C for later use.
Frozen cell pellet was resuspended in a lysis buffer containing 50 mM HEPES, 250 mM NaCl, 20 mM imidazole, 0.2 mM TCEP, and 10% glycerol, pH 7.5. Cell lysis was carried out by sonication and the resulting slurry was separated by ultracentrifugation at 185 × 103g. The supernatant was filtered through a 0.45 µm polysulfone membrane before being subjected to immobilized metal affinity chromatography. The lysate supernatant was passed over a 5 mL Ni2+-NTA column and bound proteins were eluted from the column using lysis buffer containing 300 mM imidazole. Fractions that were shown to contain His-tagged IucA by SDS-PAGE were combined and dialyzed overnight at 4 °C with TEV protease in dialysis buffer (50 mM HEPES, 250 mM NaCl, 0.2 mM TCEP, 0.5 mM EDTA, and 10% glycerol, pH 7.5). After spiking with imidazole to 20 mM, the dialyzed sample was passed over the Ni2+-NTA column for a second time. The flow-through fractions containing IucA without the His tag were combined and concentrated using a centrifugal filter before being subjected to size exclusion chromatography (SEC). The concentrated protein solution was eluted over the SEC column (HiLoad 16/60 Superdex 200, GE Healthcare Life Sciences, Marlborough, MA) using a buffer of 50 mM HEPES, 150 mM NaCl, and 0.2 mM TCEP, pH 7.5. The desired fractions, identified by chromatographic absorbance and confirmed by SDS-PAGE, were combined, concentrated, flash frozen in liquid N2, and stored at –80 °C.
High-Throughput Assay Optimization
Assay conditions were optimized in 100 µL reaction volumes in 96-well clear polystyrene microplates (NUNC Brand) and absorbance measurements were performed using a BioTek (Winooski, VT) Synergy 4 microplate reader. Initially, a ferric–hydroxamate assay was explored in which ferric iron was added to the citryl–hydroxamate product of the IucA-catalyzed reaction ( Suppl. Fig. S1 ), yielding a brown-colored chelation complex. A standard curve was constructed by combining 90 µL of acetohydroxamic acid standards with 10 µL of 10% (w/v) FeCl3·6H2O in 0.7 M HCl and measuring absorbance at 540 nm. After observing that this assay had insufficient sensitivity, an alternative MG assay was investigated ( Suppl. Fig. S1 ). 23 This assay is based on colorimetrically detecting inorganic phosphate (Pi) using an acidic solution of MG dye and ammonium molybdate. IPP was included in the reaction mixture to cleave the pyrophosphate (PPi) by-product into two equivalents of Pi and, by doing so, coupling the assay readout to IucA activity. The developing solution containing 1.0 mg/mL MG oxalate, 1.5% (w/v) ammonium molybdate, 0.15% (v/v) Tween 20, and 4.7 N sulfuric acid was prepared based on previously described methods. 24 A standard curve was constructed by combining 25 µL of this developing solution with 100 µL of sodium phosphate standards (1:4 ratio) and measuring absorbance at 637 nm. Numerous reaction parameters, including IucA concentration, substrate concentration, reaction time, and development time, were optimized in parallel using this general MG assay. The final assay conditions used in high-throughput screening are outlined below.
High-Throughput Screening Assay
For high-throughput screening, the assay volumes were cut in half for use in 384-well format (50 µL reaction solution, 13 µL developing solution). An “enzyme-initiated” protocol was carried out in which test compounds were first added to the reaction mixture before the reaction was initiated by the addition of IucA ( Suppl. Fig. S4A ). A solution containing 55.6 mM HEPES, pH 7.5, 0.11% Tween 20, 16.7 mM MgCl2, 55.6 mM hydroxylamine, 55.6 µM ATP, 55.6 µM citrate, and 0.28 U/mL IPP was dispensed (45 µL) into clear polystyrene microplates (Corning, Inc., Corning, NY) using a BioTek MicroFlo dispenser. Next, 40 nL of test compounds (10 mM in DMSO, 8 µM final concentration) was transferred from deep-well blocks to the reaction solution using a stainless steel pin tool operated by a robotic workstation (JANUS, PerkinElmer, Waltham, MA). The IucA-catalyzed reaction was initiated by adding 5 µL of 3 µM IucA in 25 mM HEPES, 75 mM NaCl, and 0.1 mM TCEP, pH 7.5. The reactions were allowed to proceed for 30 min at room temperature before being quenched by dispensing (µFill, BioTek) 13 µL of MG developing solution. After allowing the assay color to develop/stabilize for 30 min, the absorbance at 620 nm was measured (EnVision 2103 Multilabel Microplate Reader, PerkinElmer). A second, “substrate-initiated” variation of this assay was also employed in which the test compounds were allowed to preincubate with IucA in 40 µL of reaction solution (62.5 mM HEPES, pH 7.5, 0.13% Tween 20, 18.8 mM MgCl2, 93.8 mM NaCl, 0.25 mM TCEP, 0.3 U/mL IPP, and 380 nM IucA) for 15 min prior to reaction initiation with the addition of 10 µL of a substrate solution (250 µM ATP, 250 µM citrate, 250 mM hydroxylamine, and 50 mM HEPES, pH 7.5) ( Suppl. Fig. S4B ).
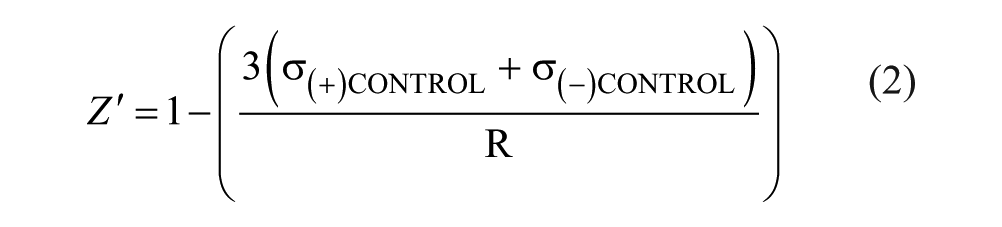
Each microplate contained positive (+) controls (DMSO vehicle only) in columns 1, 2, and 23 and negative (–) controls (either no IucA or no substrates) in column 24. Using these controls, assay performance was continually assessed by statistics including dynamic range (R) and screening window coefficient (Z′), described by Zhang and colleagues: 25
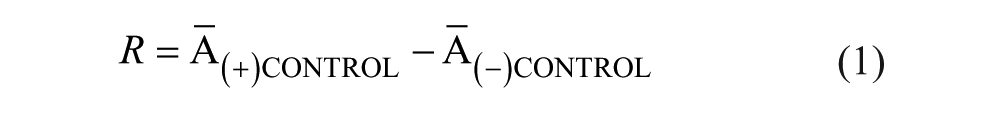
where
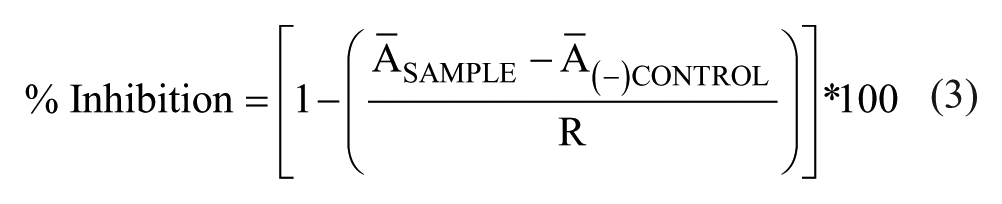
Orthogonal Luminescence Assay
The substrate-initiated IucA reaction was carried out as described above on a 100 µL scale in 96-well solid white microplates (Greiner Bio-One, Monroe, NC). Test compounds (1 µL) were added as 100× stock solutions in DMSO and allowed to preincubate with IucA in the reaction solution (80 µL) for 30 min before the reaction was initiated with the solution of substrates (20 µL). After 30 min of reaction time at room temperature, 50 µL of Kinase-Glo Plus reagent (Promega Corp.) containing luciferase and luciferin was added to the reaction mixture. The luminescence signal was allowed to stabilize for 30 min before being measured on a BioTek Synergy 4 plate reader in luminescence mode. Negative (–) controls (DMSO vehicle, minimum signal) and positive (+) controls (no IucA, maximum signal) were used to calculate percent inhibition based on light intensity in a manner analogous to the method described above. All dose–response curves of purchased compounds were fit with nonlinear regression (dose–response inhibition, variable slope) curves using GraphPad Prism 6 software (GraphPad, La Jolla, CA). IC50 values are reported ± standard error (SE) of the fitted curve.
Promiscuity AK/PK/LDH Assay
To evaluate the potential off-target effects (promiscuity) of inhibitors, a coupled AMP-NADH oxidation assay was employed ( Suppl. Fig. S1 ). 26 This assay utilizes MK, PK, and LDH to couple the production of AMP to the oxidation of NADH, which can be followed by measuring absorbance at 340 nm. A reaction mixture containing 50 mM HEPES, pH 7.5, 15 mM MgCl2, 50 mM NaCl, 0.2 mM TCEP, 0.1% Tween 20, 3 mM PEP, 500 µM NADH, and 10 U/mL MK, PK, and LDH was incubated with test compounds at room temperature for 90 min. The reaction was initiated by the addition of 500 µM AMP and 50 µM ATP. Initial reaction velocities were determined by measuring absorbance at 340 nm every 10 s for 10 min. Negative (–) (no AMP/ATP) and positive (+) controls (DMSO vehicle) allowed percent inhibition to be calculated based on initial reaction velocity in a manner analogous to the method described above.
Thermal Shift Assay
A florescence-based thermal shift assay was carried out to determine if inhibitors were able to bind and thermally stabilize IucA. 27 Samples (30 µL; 25 mM HEPES, 75 mM NaCl, and 0.1 mM TCEP, pH 7.5) were prepared containing 10 µg of IucA, 5× SYPRO Orange dye, and various concentrations of inhibitors in 96-well PCR plates (Stratagene, San Diego, CA) sealed with polyolefin film. The plates were placed in a Stratagene Mx3005P real-time PCR instrument and the temperature was increased from 25 to 99 °C over 45 min while the fluorescence intensity (λEX = 545 nm, λEM = 568 nm) was measured every 0.5 °C. Melting temperatures (TM) were determined by calculating the maximum value of the first derivative of the melting curves. When evaluating the effect of the substrates ATP (1.5 mM) and citrate (1.5 mM) on IucA thermal denaturation, a more concentrated buffer system (50 mM HEPES, 150 mM NaCl, and 0.2 mM TCEP, pH 7.5) was employed.
Results
Assay Development and Optimization
An in vitro reaction system was developed and optimized to be employed in high-throughput screening for antagonists of aerobactin synthetase IucA. Because ahLys, the native nucleophilic substrate of IucA, was not commercially available, hydroxylamine was used as a surrogate nucleophile ( Fig. 1B ). Prior studies indicated that hydroxylamine is able to react with the activated citryl–adenylate intermediate to yield a simple citryl–hydroxamate product. A reporter system was required to monitor reaction progress because none of the reaction products were conveniently directly measurable. A number of continuous and noncontinuous assay chemistries were considered for detecting each of the three products, citryl–hydroxamate, AMP, or PPi ( Suppl. Fig. S1 ).
A simple and low-cost assay based on chelating ferric iron to the citryl–hydroxamate product was initially evaluated ( Fig. 1B ). In this assay, addition of ferric iron to the reaction mixture containing the IucA-synthesized citryl–hydroxamate product led to the formation of a ferric citryl–hydroxamate chelation complex exhibiting a characteristic brown color ( Suppl. Fig. S2A ). Initial trials suggested that a relatively high concentration of substrates was required to observe a significant absorbance signal. When acetohydroxamic acid was substituted as a proxy for the citryl–hydroxamate product, a standard curve demonstrated that the ferric–hydroxamate species had a relatively low molar absorptivity (~1100 M−1 cm−1) ( Suppl. Fig. S2A ). In order to maximize the dynamic range of the assay, it was estimated that the reaction would require the substrates ATP and citrate in the low millimolar range, well above the KM values reported for IucA with these substrates (ATP, 130 ± 30; citrate, 180 ± 30 µM). 20 Because performing the reaction with saturating substrates would likely hamper our ability to identify enzyme inhibitors competitive with these substrates, we explored alternative assays with greater sensitivity.
An MG assay was evaluated for its potential to be a more sensitive alternative.23,24 In this assay, IPP was used to cleave the PPi by-product into two equivalents of Pi ( Fig. 1B ). The phosphate in the reaction mixture was then reacted with MG and ammonium molybdate under acidic conditions to form a green-colored MG–phosphomolybdate species, which was measured colorimetrically at 620–640 nm ( Suppl. Fig. S2B ). A standard curve prepared using sodium phosphate standards indicated that the MG–phosphomolybdate complex had a much higher molar absorptivity (~94,000 M−1 cm−1) than the ferric citryl–hydroxamate complex ( Suppl. Fig. S2C ).
With the MG-based assay shown to have superior sensitivity, the in vitro IucA reaction system was optimized for subsequent use in high-throughput screening ( Suppl. Fig. S3 ). A number of variables were evaluated, including reaction time, IucA concentration, substrate concentration, and development time. Due to the increased sensitivity of the MG assay, the reaction could be carried out with the concentration of ATP and citrate below their KM values, making the assay sensitive to inhibitors competitive with these substrates ( Suppl. Fig. S3A ). Concentrations greater than 50 µM exhibited a tendency for the MG–phosphomolybdate complex to precipitate and were therefore avoided in subsequent assay optimization. The reaction progress curve plateaued around 30 min ( Suppl. Fig. S3B ). To balance maximizing assay signal while maintaining sensitivity toward marginal enzyme inhibitors, a reaction time of 30 min was selected. An IucA concentration of 200–300 nM achieved a good dynamic range, while remaining insignificant compared with the test compound concentration (8 µM) ( Suppl. Fig. S3B,C ). Finally, following the addition of the development solution containing MG and ammonium molybdate, the time required for absorbance signal stabilization was dependent on IucA concentration and the amount of reaction product produced ( Suppl. Fig. S3C ). Thorough mixing of the assay solution was observed to expedite signal stabilization.
High-Throughput Screening
After optimizing assay parameters in low-throughput format, an initial automated high-throughput pilot screen was carried out. First, the assay volumes were cut in half for use in 384-well microplates (
Suppl. Fig. S4
). Next, test plates of positive (no test compounds) and negative (no IucA) controls were set up to ensure adequate dynamic range and reproducibility when utilizing automated dispensing. Finally, a pilot screen consisting of three commercially available collections totaling 4400 bioactive compounds was performed (
Fig. 2
and
Table 1
). Systematic fluctuation in the background signal periodic over two consecutive microplates was observed during the pilot screen. The fluctuation was traced to very minor dispensing oscillations, which were only appreciable due to the high sensitivity of the assay. In spite of these oscillations, assay performance appeared sufficient (average Z′ = 0.6 ± 0.1) to confidently identify assay inhibitors. By inspecting the scatterplot in
Figure 2
, 14 compounds exhibiting assay inhibition were identified and selected for follow-up testing to verify their observed activity. Of the 14, 5 compounds displayed a significant level of dose-dependent inhibition starting at 10 µM (
Suppl. Fig. S5
). Unfortunately, each of the five significantly active compounds contained a selenium or mercury atom (
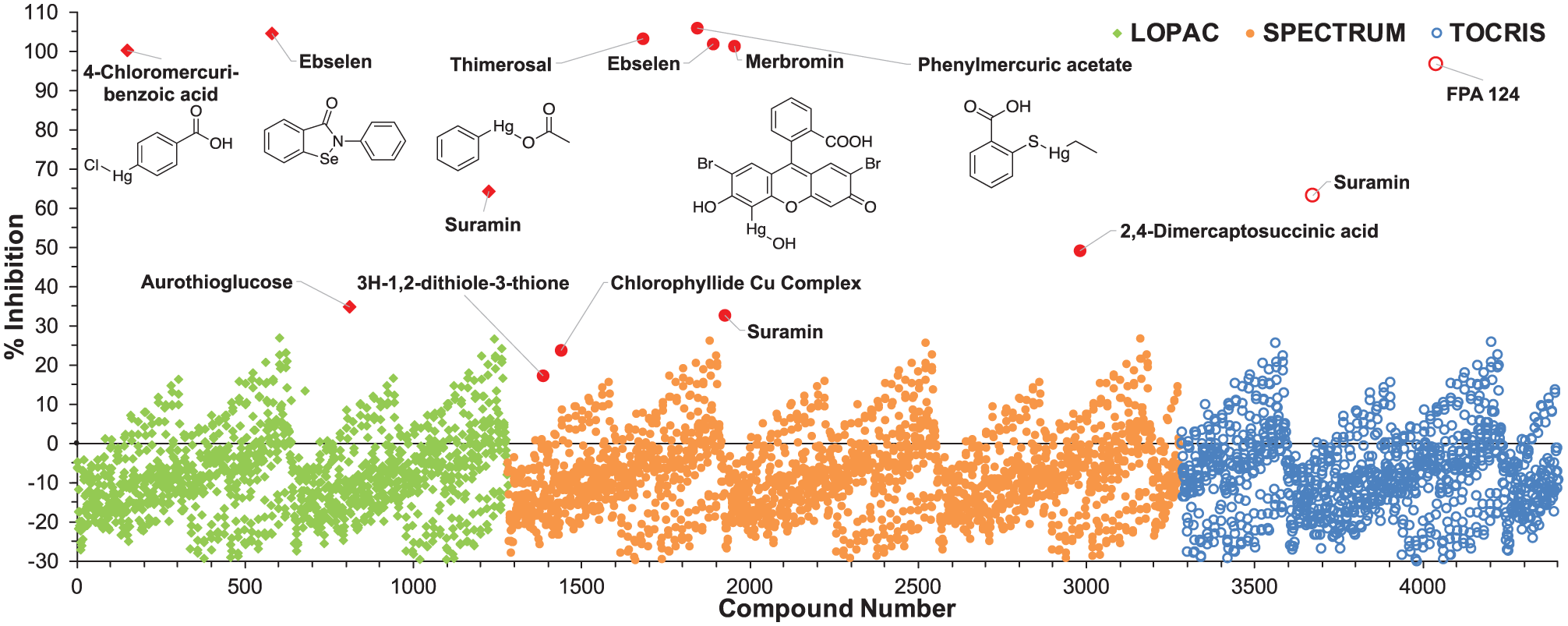
Scatterplot of the initial pilot screen of 4400 bioactive molecules from three smaller chemical libraries. Hits are highlighted in red and labeled with the compound name. The chemical structures of the five most potent compounds in follow-up testing are also included.
Statistical Summary of Screening Campaigns.
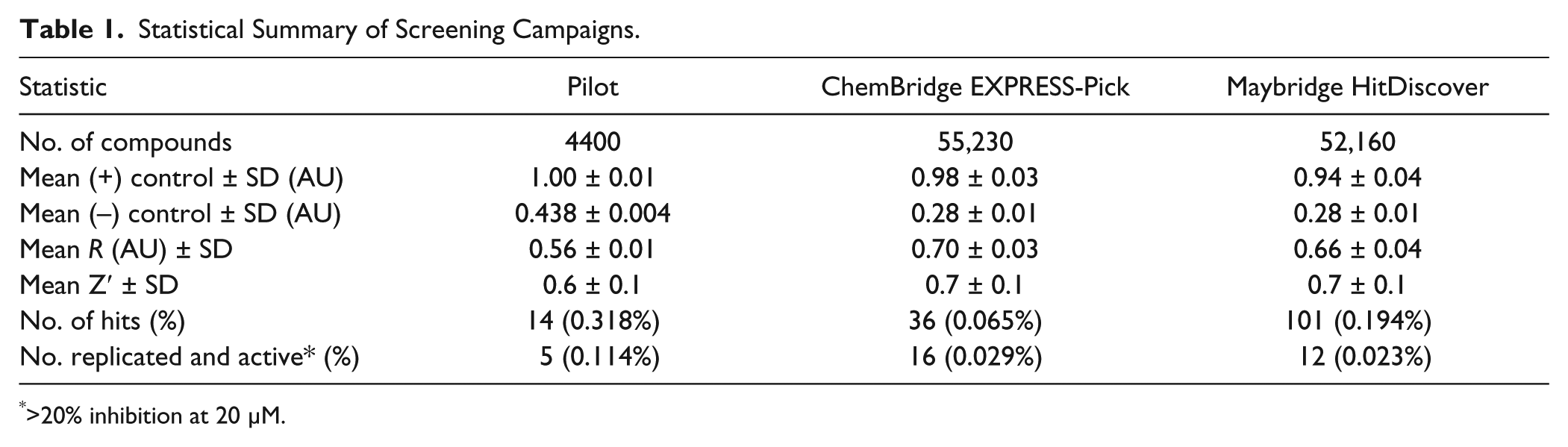
>20% inhibition at 20 µM.
We expanded our screening to encompass two larger diversity-oriented commercial libraries totaling ~110,000 compounds, seeking to discover a suitable lead for probe development ( Table 1 ). When screening the CB EXPRESS-Pick library, we used the enzyme-initiated assay protocol identical to that of the pilot screen. From this library, 36 (0.065%) hits were identified and rescreened; 16 compounds reproduced the inhibitory activity in follow-up assessment ( Fig. 3 ). The relatively low number of hits in this library led us to seek ways to adjust the assay protocol in a manner that would increase the hit rate. The protocol was modified so that test compounds were allowed to preincubate with IucA for 15 min prior to reaction initiation with a solution of substrates ( Suppl. Fig. S4B ). In this substrate-initiated protocol, slow-binding inhibitors had the opportunity to interact with IucA before the reaction was initiated. When employing this protocol for the MB HitDiscover library, the number of hits increased (101, 0.194%), although it is unclear if this was directly the result of the protocol change. Significant dose-dependent inhibition was replicated for 12 of the 101 hits ( Fig. 3 ). Across both libraries, a total of 28 compounds were identified with reproducible >20% inhibition at 20 µM.
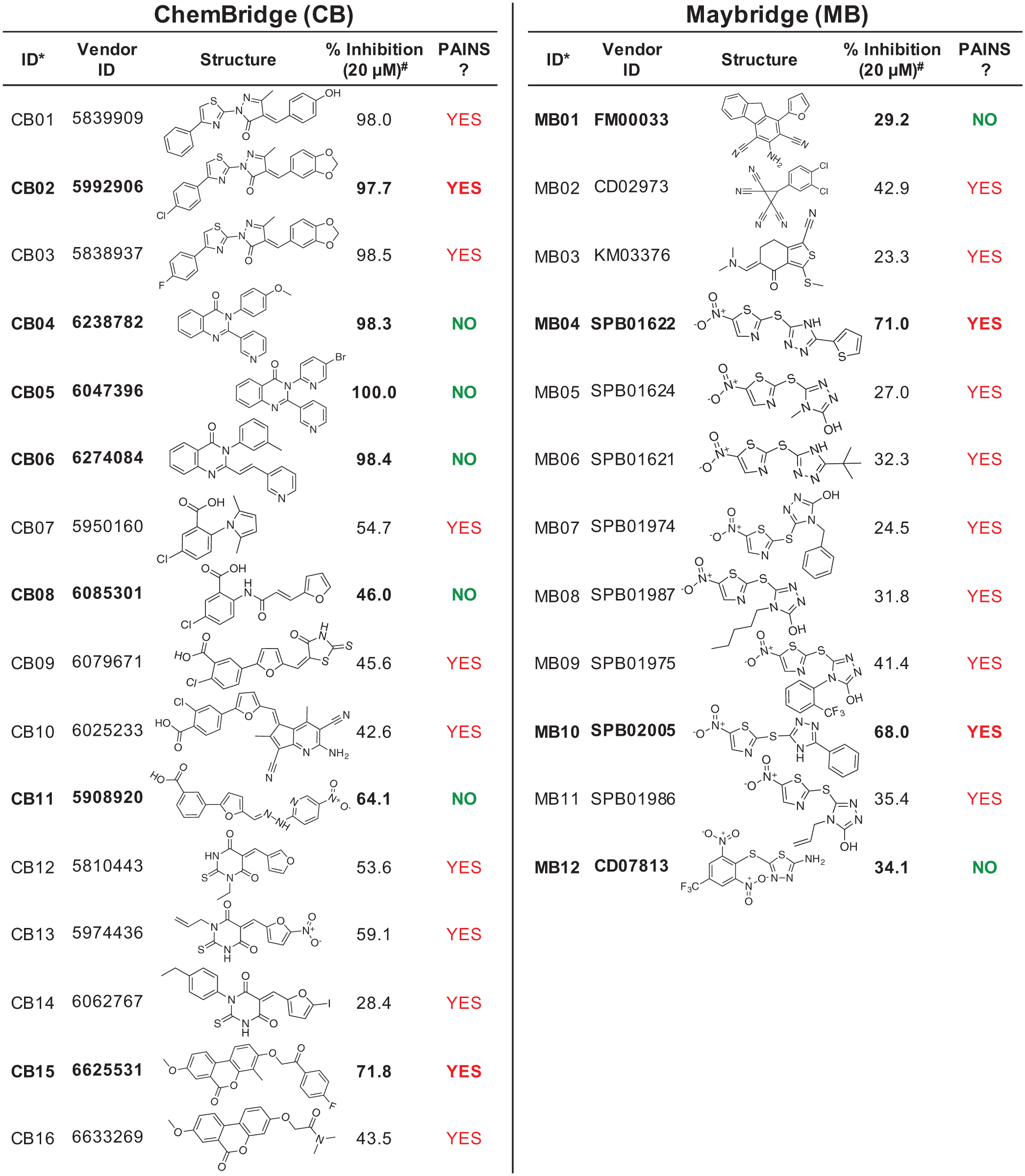
Compounds with reproducible inhibition from the CB and MB collections. Twenty-eight compounds were identified from the primary screen and showed reproducible inhibition in replicate measurements. #Percent inhibition is shown as the mean of duplicate measurements. Purchased compounds are shown in bold and were used in dose-dependent analysis.
Preliminary In Vitro Assessment of Purchased Hit Compounds
The 28 compounds with reproducible assay activity were examined to better understand their in vitro behavior ( Fig. 3 ). First, the chemical structure of the compounds was inspected for the presence of known pan-assay interference compounds (PAINS) motifs. Twenty-two of the 28 compounds were flagged as potential PAINS by an automated PAINS substructure filter. 28 Because these PAINS predictions are based on limited empiric data, we decided to proceed with further experimental evaluation against our target enzyme. 29 However, we remained cautious of the potential for inhibitor activity resulting from a mechanism other than binding site engagement.
Eleven compounds were purchased for further evaluation based on availability and chemical relatedness. The activities of the 11 purchased compounds were first retested in a dose-dependent manner using the primary substrate-initiated MG assay employed for screening the MB library (
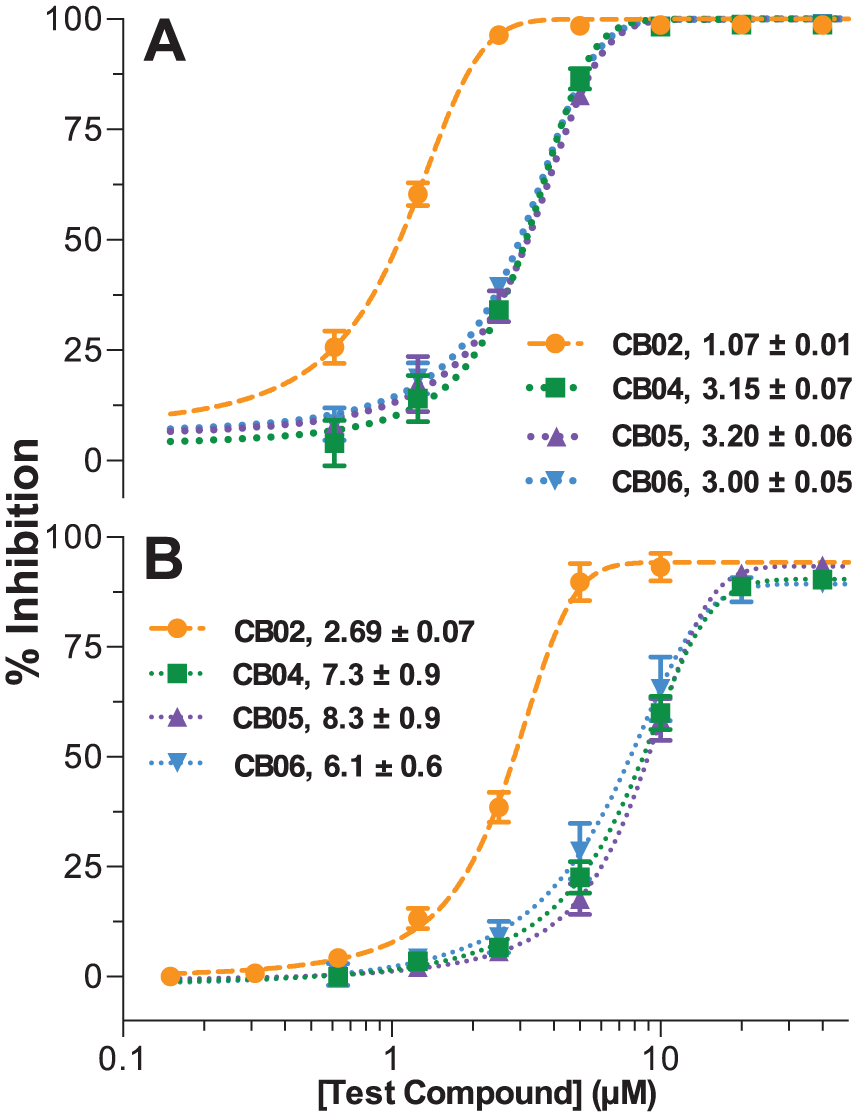
Dose–response of the most potent purchased compounds employing the (
To validate IucA-specific inhibition observed with the primary MG assay chemistry, an orthogonal luminescence-based secondary assay was employed to reevaluate eight of the most active purchased compounds (
Initial Investigation of Inhibitory Mechanism
A simple biophysical assay was then employed to determine if inhibitor–IucA binding could be detected. To this end, a fluorescence-based thermal shift assay was utilized to investigate if the purchased test compounds were able to influence the thermal stability of IucA. 27 The addition of substrates (ATP, and the combination ATP and citrate) resulted in a reproducible +2–5 °C shift in the melting temperature (TM) of IucA ( Suppl. Fig. S8A ). Knowing that ligand binding could thermally stabilize IucA, the assay was also carried out with the purchased hit compounds to probe if any could produce a similar positive thermal shift. The IucA melting curves with purchased hit compounds illustrated that none significantly shifted the melting temperature of IucA in a positive direction at concentrations up to 20 µM ( Suppl. Fig. S8B ). However, a dose-dependent increase in baseline fluorescence was observed with CB04-6 and MB12, potentially indicating a direct hydrophobic interaction with the fluorescent dye or partial denaturation of IucA.
Cognizant of the concerns raised by the PAINS analysis, further experiments were carried out to determine if these compounds were inhibiting IucA via a mechanism independent of target binding site engagement. Common characteristics of undesirable inhibitory mechanisms in biochemical assays include time dependence, sensitivity to detergent, sensitivity to enzyme concentration, and promiscuity toward unrelated enzymes.30,31 Because detergent was already included in the primary screening assay, this characteristic was not reevaluated. Only the purchased compounds with a relatively high level of dose-dependent activity, namely, CB02 and CB04–6, were evaluated further. Also, because CB04–06 shared the same quinazolinone scaffold and exhibited equivalent potency, for the sake of efficiency and conserving limited quantities of CB04 and CB05, conclusions from experiments employing CB06 were presumed applicable to all three.
When CB02 and CB06 were allowed to preincubate with IucA for 1 h prior to reaction initiation, they showed increased apparent potency compared with reactions performed without preincubation ( Fig. 5A , B ). Moreover, when these compounds were added to reactions containing increasing concentrations of IucA (20, 200, or 2000 nM), their apparent potency was reduced ( Fig. 5C , D ). Finally, the potential of CB02 and CB06 to inhibit unrelated enzymes was assessed using the coupling enzymes from the AMP-NADH oxidation assay ( Suppl. Fig. S1 ). 26 This assay contained three enzymes (MK, PK, and LDH) that could theoretically be targeted by the test compounds. Following preincubation with CB02 and CB06 for 90 min, a dose-dependent reduction in the overall reaction velocity was observed for concentrations above 6.25 and 12.5 µM, respectively ( Suppl. Fig. S9 ).
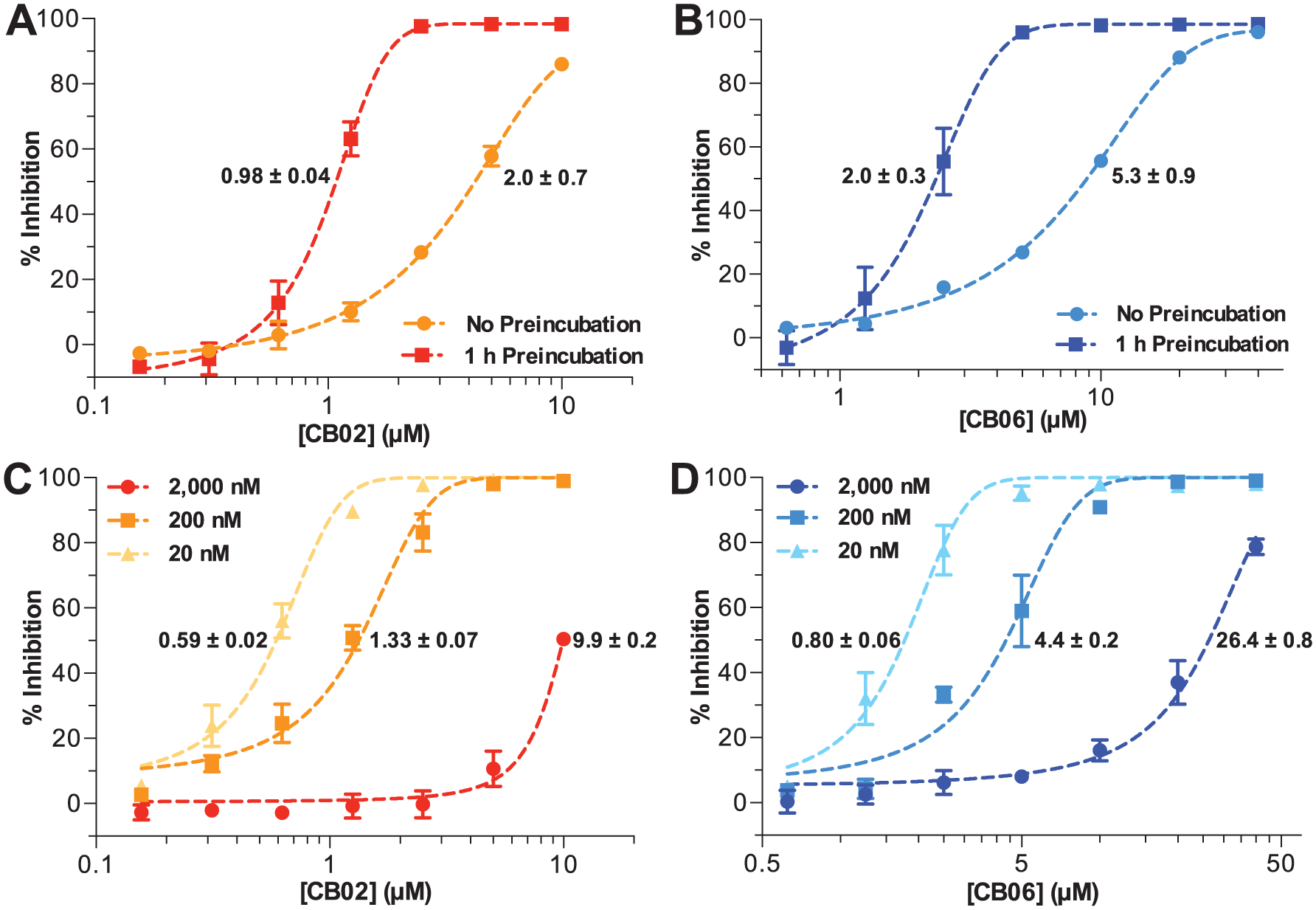
(
Discussion
We report the development of a high-throughput screening assay for inhibitors of the NIS synthetase IucA from a clinically isolated strain of hvKP. The assay was optimized for substrate and enzyme concentrations, reaction and development times, and employing a surrogate nucleophile in place of the native ahLys substrate. Following assay development and optimization, we report here the screening of multiple small-molecule libraries, as well as preliminary biochemical and informatic analyses of identified active compounds.
The assay was robust when miniaturized to 384-well plate format, enabling the screening of initial pilot libraries of a few thousand compounds. In the pilot screen, the assay displayed satisfactory performance with an average Z′ factor of 0.6, despite the average negative control (0.44 AU) displaying higher background signal than expected ( Table 1 ). The assay proved capable of identifying enzyme inhibitors, with five compounds displaying significant dose-dependent inhibition. Ebselen was identified as an inhibitor from both the LOPAC and Spectrum Collection. Known as a cysteine modifier, ebselen is a potent covalent inhibitor of a number of thiol-dependent enzymes. 32 While IucA is not predicted to rely on a cysteine thiol for its catalytic activity, general nonspecific electrophilic modification of the enzyme could be responsible for the apparent activity of ebselen. In addition, four organomercury compounds were also identified in the initial screen. Merbromin, thiomersal, and phenylmercuric acetate were once widely employed in antiseptics, preservatives, or disinfectants, while 4-chloromercuribenzoic acid is a well-known thiol-reactive enzyme inhibitor.33,34 Like ebselen, these organomercury compounds likely inhibited IucA via nonspecific electrophilic reactivity. Despite uncovering inhibitors with probable nonideal mechanisms of action, this limited preliminary screen proved that the assay was capable of picking out inhibitors.
Two larger diversity-oriented collections were screened with the goal of identifying a more suitable lead for probe development. First, screening the 55,230-compound CB EXPRESS-Pick library (average Z′ = 0.7 ± 0.1) turned up 36 (0.07%) compounds with apparent inhibitory activity. Preincubation of test compounds with the target is reported to increase the chances of identifying slow-binding inhibitors. 35 Therefore, the 52,160-compound MB HitDiscover library was screened (average Z′ = 0.7 ± 0.1) using a second, substrate-initiated protocol in which IucA and test compounds were preincubated for 15 min prior to reaction initiation with substrates. The use of this experimental protocol resulted in a significantly increased hit rate (101, 0.19%), but the majority (89 out of 101) were not replicated in follow-up testing. The underlying mechanism behind the increased noise of the substrate-initiated protocol is unknown. In the end, a similar number of inhibitors with significant replicated activity were identified in both libraries (CB = 16 and MB = 12).
Many (22/28) of the active compounds were flagged for containing predicted PAINS motifs. In order to experimentally evaluate the empiric predictions, 11 compounds were purchased for further assessment. Compounds CB08, CB11, and CB15 only displayed significant inhibitory activity when tested using the original enzyme-initiated protocol. Given the resemblance of CB11 to the antibiotic nitrofurantoin, we hypothesized that CB11 may rely on redox activity to inhibit IucA. Its differential activity could be due to differences in TCEP exposure between the two protocols. Furthermore, this compound has also been reported as an inhibitor in a number of other reported high-throughput biochemical screens.36–39 The underlying mechanism behind the differential activity of CB08 and CB15 remains unclear.
Of the 11 purchased compounds, 4 displayed substantial dose-dependent inhibition of IucA in both the primary MG and the secondary luminescence assays. The most potent compound, pyrrolone-containing CB02, displayed IC50 values of 1–3 µM. CB04–06, which all shared a quinazolinone scaffold, also displayed promising dose-dependent inhibition with IC50 values of 3–8 µM. Comparable inhibitory activity in the primary and secondary assays, which utilized distinctive chemistries coupled to unique products of the IucA-catalyzed condensation, suggested that the compounds were acting as target enzyme inhibitors. However, we remained cautious, particularly toward CB02, which was highlighted for containing a PAINS substructure motif.
Subsequent experiments were carried out to better characterize the in vitro behavior of the pyrrolone and quinazolinone compounds. The substrate ATP, and the combination of ATP and citrate, induced a positive shift in the thermal denaturation of IucA. In contrast, none of the quinazolinones were observed to stabilize IucA by the thermal shift assay. However, melting curves with these compounds exhibited a dose-dependent increase in the baseline fluorescence, potentially a consequence of direct hydrophobic interaction with the fluorescent dye, or the compounds inducing partial unfolding of IucA. Given the recent attention that promiscuous aggregating inhibitors have garnered in the context of biochemical screening, we wanted to evaluate compound aggregation as a potential mechanism at an early stage in the discovery process.28,30,31 Testing of CB02 and CB06 revealed potential evidence of aggregation-based inhibition, including preincubation time dependence, enzyme concentration sensitivity, and promiscuity toward other enzymes. In this last assay, the promiscuity of the inhibitors was evaluated with an extended preincubation time (90 min) and with three possible enzyme targets. Inhibition was observed in this assay; however, a cumulative IC50 (50% inhibition against the three coupling enzymes) was approximate 20× higher for both CB02 and CB06 when compared with the preincubated inhibition against IucA.
To provide additional context to the apparent antagonism of CB02 and CB04–06 against our target enzyme IucA, we also examined their reported activity in publicly available screening results. The pyrrolone compound CB02 (CID 5345903) was reported active in two out of eight unique bioassays in the PubChem database, including against HIV-1 RNase H and CDC25B-CDK2/cyclin A interaction. In contrast, no activity was reported for the quinazolinones CB04–06 (CIDs 5049702, 4359529, and 5729808), which were employed in seven, two, or one unique assay deposited in PubChem, respectively.
Further examination of the literature revealed that the pyrrolone and quinazolinone compounds have also been previously reported as inhibitors in high-throughput biochemical screening projects. CB04 and CB05 were both identified as potent inhibitors of bacterial IspE kinase and Plasmodium falciparum glucose-6-phosphate dehydrogenase 6-phosphogluconalactonase in two disparate biochemical screening campaigns.36,40 They were characterized as irreversible inhibitors with apparent potency dependent on target preincubation time. Furthermore, structure–activity relationship (SAR) investigation of the original lead was ambiguous and failed to identify a more potent analogue. The quinazolinone CB06 was identified in a third unrelated high-throughput biochemical screen as an inhibitor of human RAD51 recombinase in double-strand DNA repair. 41 In contrast to CB04 and CB05, SAR analysis of CB06 revealed that RAD51 inhibition was sensitive to minor structural modifications, and it did not inhibit the E. coli homologue RecA. The pyrrolone compounds CB01–03 were flagged for containing a PAINS motif, and all were previously identified as inhibitors in high-throughput biochemical screens against human and Yersinia fatty acid synthases.39,42 Thus, while the prior reports suggest that these compounds can antagonize other target enzymes in alternate assay environments, they do not rule out their potential utility as a lead for blocking aerobactin biosynthesis.
With aerobactin shown to play a critical role in the evolution of a novel hypervirulent pathogen (hvKP), we sought to develop a platform for identifying inhibitors of this critical virulence factor. Multiple lead compounds were identified against IucA following the development of a high-throughput biochemical screening assay. Inhibitors with activity against IucA by two orthogonal assays exhibited in vitro potency in the low-micromolar range. Initial follow-up experiments on the most potent inhibitors revealed some concerning in vitro characteristics consistent with nonspecific enzyme interference mechanisms. Furthermore, any intracellular inhibitors will also face the challenge of cellular penetration, which is a particularly challenging problem for targeting Gram-negative bacteria. The use of more diverse libraries, including natural products, or those that may incorporate chemical features known to enable antibiotic accumulation, 43 may help exploit this novel antivirulence target in an increasingly problematic pathogen.
Supplemental Material
Supp_Info_Inh_Aerobactin_Syn_IucA_Bailey_et_al – Supplemental material for Development of a High-Throughput Biochemical Assay to Screen for Inhibitors of Aerobactin Synthetase IucA
Supplemental material, Supp_Info_Inh_Aerobactin_Syn_IucA_Bailey_et_al for Development of a High-Throughput Biochemical Assay to Screen for Inhibitors of Aerobactin Synthetase IucA by Daniel C. Bailey, Brian P. Buckley, Mikhail V. Chernov and Andrew M. Gulick in SLAS Discovery
Footnotes
Acknowledgements
We thank Dr. Courtney Aldrich for helpful insights regarding hit compound evaluation. We thank Mr. Eric Drake for helpful discussions concerning assay design and development. Finally, we thank Dr. Thomas Russo for valuable dialogues and insights on hvKP.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the National Institutes of Health (AI-116998 to A.M.G) and pilot studies support from the Buffalo Clinical and Translational Research Center (NIH grant UL1TR001412 [Timothy F. Murphy]). D.C.B. was supported by NIH training grant T32-AI007614 (Laurie K. Read).
Supplemental material is available online with this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.