
Abstract
The establishment of mammalian cell lines reliably expressing G-protein-coupled receptors (GPCRs) can be a tedious and often time-consuming process. A strategy has been developed to allow the rapid production of such cell lines. The first step of this approach was the generation of a specialized master cell line, characterized by optimized stable expression of a membrane-bound reporter protein. In the second step, this reporter gene was exchanged for that of the GPCR of interest by a DNA recombinase “cut-and-paste” engineering step. It has been demonstrated that the resulting GPCR cell lines inherit the advantages of the master cell line, expressing the GPCR in a homogeneous and stable manner. The case studies presented demonstrate the functionality of the established GPCR cell lines, and most important, because of the highly efficient integration event, these recombinant GPCR-expressing cell lines were generated within a timeframe of 2 to 4 weeks. The advantages of this cut-and-paste approach versus other strategies such as Flp-In or Jump-In are compared.
Keywords
Introduction
T
Upon modulation by extracellular ligands, these cell surface receptors mediate the transduction of extracellular stimuli into intracellular signals. The hydrophobic core composed of seven-transmembrane α-helices (7TM) is common to all GPCRs. 2 This large class comprises approximately 800 different receptors that are grouped in three families (classes A, B, and C).
In general, the generation of cell lines stably expressing a transgene requires transduction of the host cell line and subsequent integration of the expression cassette into the host cell genome. This leads to a random distribution of integration sites. 3 It is a fact that most genomic loci do not support high or consistent transgene expression. Both the level and stability of transgene expression are strongly dependent on the genomic surroundings of the integration sites. This “position effect” renders the generation of stable cell lines a cumbersome and often lengthy task involving extensive screening to identify those cell clones that display the desired properties. 4
The recombinase-mediated cassette exchange (RMCE) strategy was introduced to overcome this limitation of cell line engineering (for review, see Wirth et al. 5 ). RMCE can be best described as a two-step molecular “cut-and-paste” mechanism. 6 In the first step, a reporter gene cassette is randomly integrated into a host cell line of interest. The expression of the reporter gene serves as a marker that permits screening for the desired expression characteristics (e.g., high and stable expression). Once isolated and confirmed, those “tagged” clones function as master cell lines. A prerequisite for the use of such a master cell line is a single-copy integration of the tagging cassette. With the help of DNA recombinases such as the Cre, 7–9 the Flp recombinase, 10–12 or the phiC31 integrase 13,14 the tagged genomic loci of the master cell lines can be recycled by the integration of any gene of interest. Because the recombinase-driven cassette exchange itself is a highly site-specific event, all positive expression characteristics of the master cell line are transferred to the resulting subclonal producers.
The aim of the study presented here is to employ the RMCE strategy for the establishment of GPCR-expressing cell lines. The main application area of these cell lines is the drug discovery process. Therefore, the cell lines should meet the following requirements: (1) consistent expression, (2) stable expression without a need of persistent use of selection agents, and (3) sufficient expression of the receptors to monitor pharmacological responses. The most significant feature of the approach described here is that the GPCR-expressing cell lines should be rapidly established (within 2–4 weeks) while still showing the above-mentioned properties.
The RMCE strategy has been proven useful for the flexible generation of various production cell lines expressing, for example, antibodies or retroviral vectors. Although the principle itself can be adapted to virtually any desired gene, it has become clear that there can be no universal master cell line. This is because the expression characteristics of a cell line are mainly influenced by the position effect, the design of the expression cassette, and last but not least the transgene itself. 15 In approaches described previously, cytosolic reporters (green fluorescent protein [GFP], lacZ) were used for the establishment of the master cell lines. 11,12
The present study follows the hypothesis that a master cell line established through expression of a protein that recapitulates the complex maturation process of a GPCR is better suited to stably and consistently express the GPCR of choice. Therefore, novel Chinese hamster ovary (CHO) master cell lines have been generated dedicated for GPCR expression. The key features of these master cell lines are (1) the selection for stable expression of membrane-bound proteins and (2) a highly efficient targeting process that ensures site-specific cassette exchange in a significant number of master cells. The RMCE process used was extremely precise with 100% positive recombination events in the selected clones. 11,12 However, previously described master cell lines gave rise to few recombined clones, making RMCE a rare event. Improvement in the frequency of the RMCE process would lead to more clones. In addition, because of their isogenic nature, the clones can be pooled, enabling a further shortening of the time needed for establishment of a stable line from 4 to 2 weeks.
Materials And Methods
Plasmids
The vector for tagging chromosomal sites harbors a truncated version of the nerve growth factor receptor (NGFR). This gene mutant acts as a reporter gene and therefore lacks the internal domain of the NGFR to avoid any signaling from this receptor. In addition, the tagging vector harbors the selection marker hygromycin phosphotransferase, thymidine kinase whose expression is mediated by a poliovirus internal ribosomal entry site (IRES). Both genes are driven by an SV40 promoter. The cassette is flanked by a wild-type FRT site and F5 spacer mutant FRT site, 16 followed by an ATG-deficient neomycin phosphotransferase gene (neoR).
All RMCE cassettes coding for the GPCRs of interest are also driven by a SV40 promoter and are flanked by FRT wild-type and the F5 FRT mutant sites. The vectors carry an IRES element and an ATG start codon upstream of the FRT F5 mutant site to complement the inactive neoR gene upon successful RMCE process. A successful RMCE process was verified by a PCR employing primers CGTCAAGAAGGCGATAGAAGGC (NeoRev) and ccatgggacgctagttgtgaa (Polio1), which bind in the ATG-deficient neomycin (from the master cell line) and in the IRES from the targeting plasmid, respectively. For monitoring the modulation of the GPCRs, the reporter construct pGL4.29 (Promega, Madison, WI) was used. pGL4.29 harbors a hybrid promoter composed of a minimal promoter with CRE binding elements and drives the expression of the reporter gene luciferase.
Mammalian cell culture and transfection
CHO-K1 cells (ECACC, Salisbury, UK) were cultivated at 37 °C in a humidified atmosphere with 5% CO2 in Ham-F12 medium (GIBCO, Carlsbad, CA) containing 10% fetal calf serum and 8 mM L-glutamine. For transduction of the CHO-K1 cells, the respective vectors were electroporated using a standard protocol (Amaxa AG, Cologne, Germany; Nucleofector Kit V). For the tagging procedure, 1 × 106 cells were transfected with the NGFR expression plasmid. The transduced cells were selected with medium supplemented with hygromycin B (150 U/mL). The RMCE process in the CHO-K1 master cell line was facilitated by a co-transfer of Flp recombinase-expressing vector and the targeting plasmid. 17 For the reporter assays, the pGL4.29 was electroporated into the respective GPCR-expressing cell lines and as a control also into the master cell line. Cryopreserved “Assay-Ready” reagent for high-throughput analysis was prepared in multilayer bioreactors (Corning Cellstack; Corning, Inc., Corning, NY). Cryopreservation was carried out using rate-controlled freezer (Planer, Sunbury-on-Thames, Middlesex, UK).
Cellular analysis
FACSCalibur and FACSVantage SE (Becton Dickinson, Franklin Lakes, NJ) were used for flow cytometry analysis and cell isolation. The analysis and sorting of the NGFR-expressing cells was done as previously described. 18 Briefly, the transduced cells were incubated for 30 min with a primary mouse monoclonal antibody against NGFR (Becton Dickinson) at room temperature. Afterwards, the cells were washed twice with phosphate-buffered saline (PBS) supplemented with 2% fetal bovine serum (PBS*). As a secondary antibody, an anti-mouse PE-conjugated antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) was used (30 min at room temperature). Afterwards the cells were washed with PBS*, stained with propidium iodide (50 µg/mL) to exclude dead cells from the analysis, and then analyzed by flow cytometry. For the analysis of the ADRB2-expressing cells, a fluorescently labeled ligand specific for ADRB2 (CellAura, Nottingham, UK) was used. The cells were incubated with the ligand (0.4 nM) at room temperature for 5 min. Afterwards, the cells were washed with PBS* and analyzed by flow cytometry or by immunofluorescence. All chemicals used in this study were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. For determination of the reporter plasmid activity, a luciferase assay was performed as detailed in Nehlsen et al. 19 Briefly, cell lysates were created by repeated freeze-thaw cycles. An aliquot of the protein lysate was mixed with reaction buffer (25 mM glycylglycine, 15 mM MgSO4, 1 mM adenosine triphosphate [ATP], pH 7.8) and luciferin (Promega). The resulting luciferase activity was measured in a luminometer (Lumat LB9507; Berthold, Bad Wildbad, Germany) and corrected for protein content (RLU/µg protein).
High-throughput analysis
The analysis of the pharmacological characteristics of the muscarinic M3 receptor cell line was performed using the FLIPRTETRA system (Molecular Devices, Wokingham, Berkshire, UK). Cells in continual culture were plated into Corning CellBIND black/clear base 384-well plates at 10 000 cells/well in 50 µL Ham F-12, left on the bench for 60 min at room temperature, and then grown overnight at 37 °C, 95% humidity, and 5% CO2. Dye loading buffer was prepared by dissolving the contents of one vial of FLIPR Calcium 5 Reagent completely with a final volume of 20 mL Hank’s balanced salt solution, 20 mM HEPES, and 2.5 mM probenecid adjusted to pH 7.4. Cell plates were removed from the incubator, growth media were removed, and 50 µL dye loading buffer was added to each well. Dye-loaded plates were incubated for 45 min at 37 °C, 5% CO2 and allowed to cool to room temperature for 15 min prior to reading on the FLIPRTETRA system (ex: 470-495 nm; em: 515-575 nm). Plates were not washed after dye loading. Compounds were added as 12.5 µL of a 5× ligand stock. For IC50 determinations, an EC80 concentration of acetylcholine was used as the standard agonist. Data analysis was performed using GraphPad Prism (GraphPad Software, La Jolla, CA) software. The data were normalized as percent change over baseline.
Results
Development of GPCR-optimized master cell lines
Following the hypothesis that the reporter protein for the tagging step should reflect the properties of the later protein of interest, a novel tagging cassette was constructed coding for the membrane-bound NGFR.
The tagging cassette was electroporated into CHO cells, and after a selection period of 14 days, the cells were analyzed and the highest yielding 10% of NGFR-expressing cells was cloned by flow cytometry ( Fig. 1 ). In total, 219 clones were cultivated for 4 weeks without selection pressure and then reanalyzed for NGFR expression. Of these clones, approximately 10% (25 clones) displayed a stable and homogeneous NGFR expression (see Fig. 1 for representative clone). Subsequently, these clones were analyzed for “targetability” (ability to perform RMCE) and long-term stability of NGFR expression (> 4 months without selection pressure). This resulted in the generation of two cell lines showing stable NGFR expression for 50 passages (more than 20 weeks). In a standard RMCE procedure, these master cell lines typically gave rise to 200–300 clones, which were approximately a 10-fold higher efficiency than in previously published reports. 11,12 Thus, these cell lines meet the above-described requirements and can serve as novel master cell lines (SCREENflex-GR1 and SCREENflex-GR2). For the subsequent experiments, SCREENflex-GR1 was used.
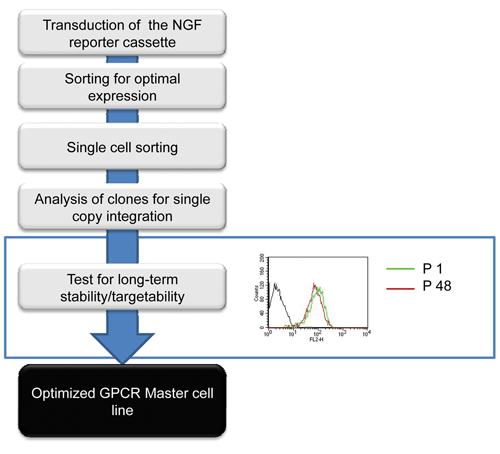
Scheme for the establishment of novel master cell lines. Flowchart describing the process for generating the novel master cell line optimized for the expression of membrane-bound proteins. CHO-K1 cells were electroporated using a nerve growth factor receptor (NGFR) expression vector to tag genomic loci supporting high-level expression of a membrane-bound protein. After transduction, cells were selected and single cells were sorted. The resulting clones were expanded for 4 weeks without selection pressure and reanalyzed for NGFR expression. The NGFR-positive clones were assessed for their capability to perform the recombinase-mediated cassette exchange (RMCE) process and for long-term stability (>4 months) of the NGFR expression. GPCR, G-protein-coupled receptor.
Establishment of various GPCR-expressing cell lines
For the generation of the GPCR-expressing cell lines, targeting vectors were cloned that allow site-specific integration into the NGFR-tagged locus of SCREENflex-GR1 ( Fig. 2A ).
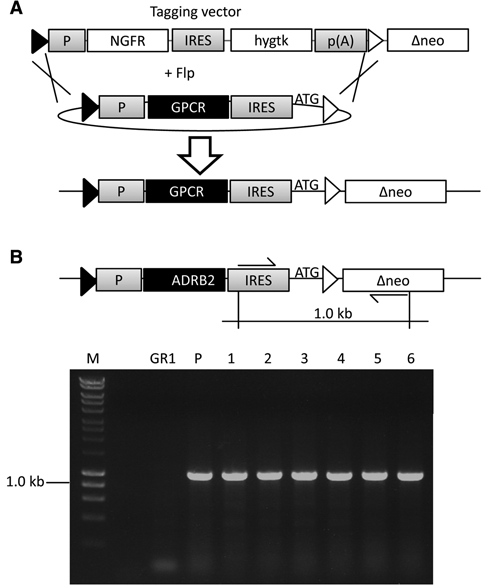
Recombinase-mediated cassette exchange (RMCE) in the novel master cell lines. (
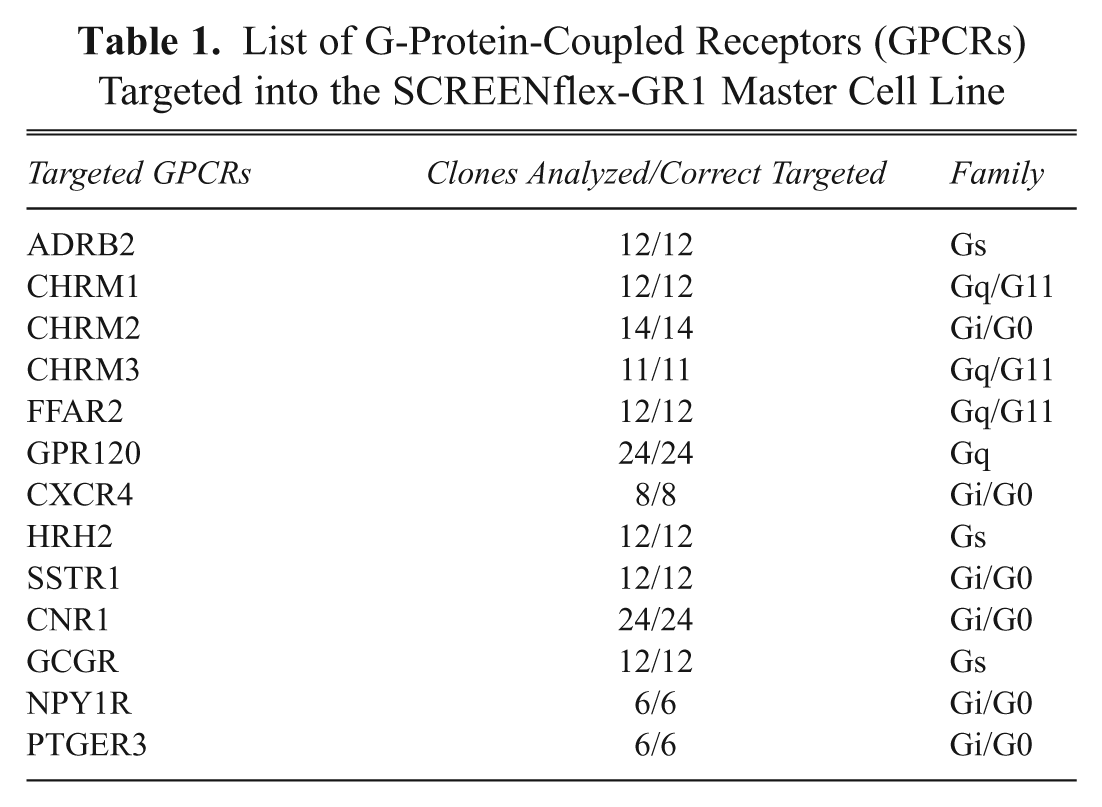
The cDNAs of 13 different GPCRs were cloned into this targeting vector ( Table 1 ) and used for RMCE experiments. After electroporation of both a flipase encoding vector and a targeting vector, selection with G418 was applied to screen for those cell clones that complemented the defective neomycin. For all GPCR targeting experiments performed, neomycin-resistant colonies were obtained within 7–10 days of selection. Clones of G418 resistant cells were randomly picked and expanded. The average time required to expand the GPCR-expressing cells (expansion to 5 Mio. cells) was within 4 weeks. G418-resistant colonies were pooled to obtain a mixture of GPCR-expressing cells. Both pools and clones were analyzed by PCR for correct cassette exchange. The expected band (1 kb) was detected for both clones and pool in all established GPCR cell lines (representative data shown for ADRB2 targetings in Fig. 2A ).
List of G-Protein-Coupled Receptors (GPCRs) Targeted into the SCREENflex-GR1 Master Cell Line
Expression of the β2-adrenergic receptor
The expression of the β2-adrenergic receptor (ADRB2) shows, as an example, the performance of the above-described procedure. ADBR2 is a prototypic and well-characterized member of the GPCR family, which falls into the class of adrenoreceptors with adrenaline being the natural ligand. 20 In parallel to the RMCE approach, recombinant ADBR2-expressing cell lines were generated by co-transfection of the ADBR2 expression plasmid and a selection plasmid into CHO-K1 wild-type cells. After selection, 18 clones were randomly picked and expanded for further analysis.
To compare the expression of the ADRB2 receptor of the targeted pool, targeted subclones, and randomly integrated clones, a fluorescent ligand was used that specifically recognizes ADBR2 receptor. The docking of the ligand was followed by fluorescence microscopy ( Fig. 3A ) as well as flow cytometry ( Fig. 3B ). The analysis demonstrated that the cell clones and the pool derived by RMCE were composed of 100% ADBR2-expressing cells, showing a homogeneous expression pattern of the GPCR. This is in line with the genetic analysis showing isogenicity of the targeted subclones. Therefore, the generated pools can be considered “pure clones.” This result is important as a pooling step could lead to heterogeneously expressing cell lines, which could complicate the analysis of further tests.
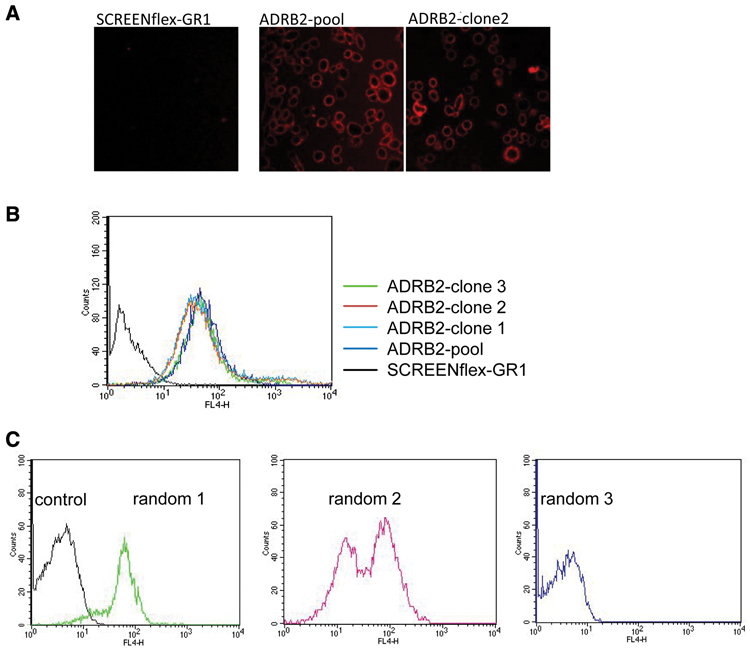
Characterization of cell lines expressing β2-adrenergic receptor (ADRB2). ADRB2-expressing cell lines were established (
The cells generated by random integration were also analyzed with the fluorescent ligand using flow cytometry. This analysis showed a different picture if compared to the RMCE approach. The 18 individual clones could be divided into three different categories concerning their expression characteristics: (1) 3 clones that expressed ADBR2 homogeneously, (2) 4 clones that showed a mixed expression of ADBR2, and (3) 11 clones that expressed no or only low amounts of ADBR2 ( Fig. 3C ). This suggests that for the generation of recombinant cell lines by random integration, a time-consuming screen for those cells that express the transgene in the desired fashion is required.
Functional characterization of targeted GPCR cell lines
Cell lines generated by RMCE were further analyzed to determine if they follow typical pharmacological responses after challenge with corresponding ligands. For this analysis, the ADRB2 cell line (see above) and also a SCREENflex-GR1-based cell line expressing the human glucagon receptor (GCGR) were included ( Table 1 ). The main downstream activity of both GPCRs is primarily coupled to the Gs pathway, leading to the stimulation of adenylyl cyclase activity. 21 Therefore, the established cell lines were investigated to determine if there was an increase in the production of cAMP upon stimulation. For this purpose, a reporter construct was used in which luciferase expression is driven by a promoter that harbors cAMP-responsive element-binding protein (CREB) binding sites ( Fig. 4 ). CREB is a downstream effector of cAMP, and therefore any change of cAMP level is indirectly measured. This expression construct was stably integrated in the established ADRB2 and glucagon receptor–expressing cell lines (clones and pool). These cells were then treated respectively with either isoprenaline, a known inducer of ADRB2, or glucagon. Both receptor cell lines exhibited a strong increase in luciferase expression upon induction with their ligands ( Fig. 4 ).
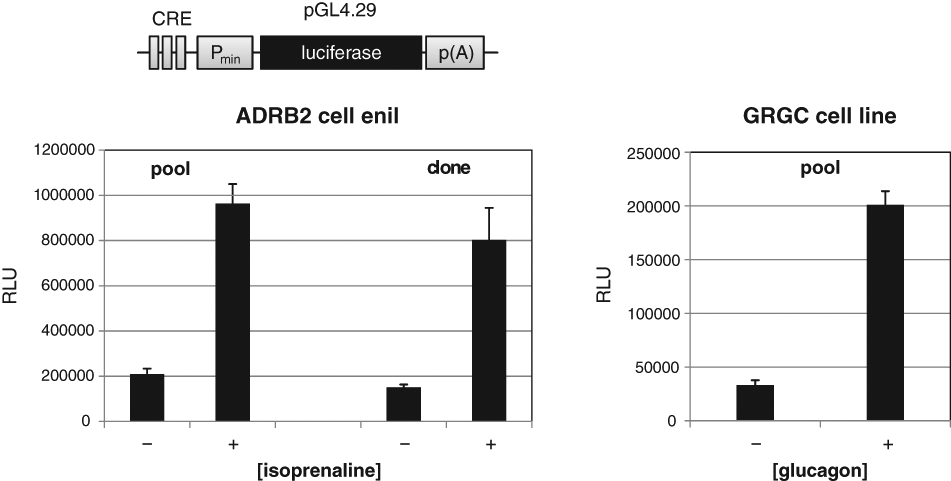
Pharmacological properties of G-protein-coupled receptor (GPCR) cell lines. The pharmacological properties of the established GPCR-expressing cell lines were assessed by the transfection of a reporter plasmid in the respective cell line. The reporter plasmid contains a hybrid promoter that is composed of CRE binding sites and a SV40 minimal promoter. The hybrid promoter drives the expression of the reporter gene luciferase. After transduction of the reporter construct, the GPCR-expressing cell lines were cultivated with and without a known agonist, and the activity of luciferase was determined. The diagrams show the response of β2-adrenergic receptor (ADRB2)–expressing cell line to isoprenaline (clone/pool) and of human glucagon receptor (GCGR) to glucagon (10−6 M each).
High-throughput screening compatibility of the cell lines established by RMCE
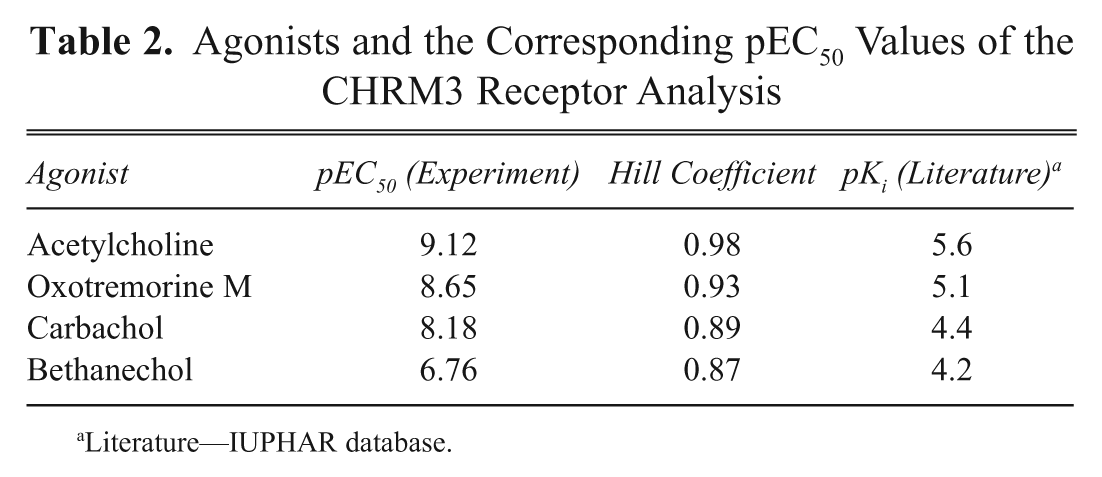
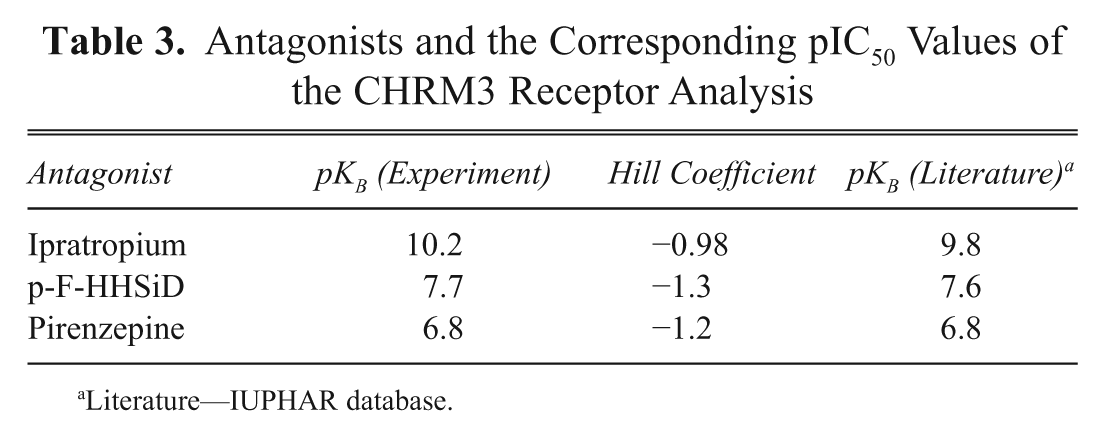
The identification of novel modulators of GPCRs is a major task of current drug discovery programs; for this purpose, large compound libraries are typically screened in a high-throughput fashion. To achieve this in an acceptable timeframe, the corresponding assays are performed in a 384- or 1536-well format. Therefore, one of the prerequisites of a GPCR-expressing cell line is the demonstration of the correct and consistent pharmacological response profile. In a pilot study, a human muscarinic receptor 3 (CHRM3) cell line was generated with the RMCE approach ( Table 1 ) and assayed in a 384-well format using a FLIPRTETRA System. In a series of experiments, four known muscarinic agonists (acetylcholine, oxotremorine M, carbachol, and bethanechol) and three known antagonists (ipratroprium, p-F-HHSiD M, pirenzipine) were used to characterize the cell line ( Fig. 5A , B ). The assay is based on an increase of cytosolic Ca2+ ions upon induction with specific ligands (due to Gq protein coupling of the respective receptor). The increase of fluorescence signal due to the binding of calcium to a calcium-sensitive dye (FLIPR Calcium 5 Assay) served as a readout. The different compounds produced the expected pharmacological response profiles ( Fig. 5A , B ), and the rank order of potency is the same for the agonists and antagonist as compared to those described in the IUPHAR database ( Tables 2 and 3 ). The values measured for the agonists differ slightly from those described in IUPHAR. This is possibly because the IUPHAR values describe affinity binding estimates, whereas the pEC50 data presented here are based on functional response. What is significant, however, is that the hierarchy of potency is the same as described in IUPHAR. The antagonist data, on the other hand, are equivalent to those in the IUPHAR database ( Tables 2 and 3 ).
Agonists and the Corresponding pEC50 Values of the CHRM3 Receptor Analysis
Literature—IUPHAR database.
Antagonists and the Corresponding pIC50 Values of the CHRM3 Receptor Analysis
Literature—IUPHAR database.
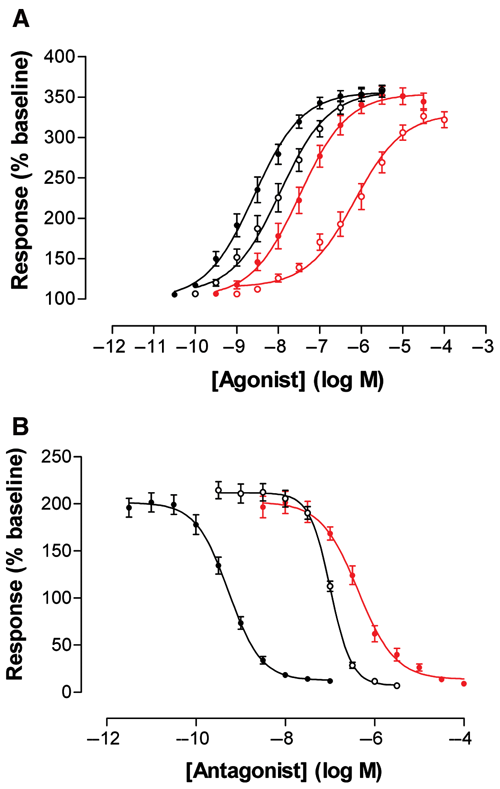
Analysis of high-throughput screening (HTS) compatibility of a CHRM3 cell line. Four known agonists (
Discussion
In the present study, the generation of novel master cell lines was verified, specifically designed for the expression of membrane-bound proteins. For this purpose, NGFR was used to screen for chromosomal integration sites that support a stable and appropriate expression of membrane-spanning proteins. The implementation of the recombinase-based cassette exchange principle allowed the reuse of such master cell lines (tagged loci). Because no laborious testing for proper expression features was necessary, all cell lines were able to be generated in parallel. This was validated by rapidly establishing 13 different GPCR-expressing cell lines with the help of the novel master cell line SCREENflex-GR1 ( Table 1 ). All cell lines were generated in a timeframe of 2 to 4 weeks; this was possible because of the high number of RMCE events. The exchange of the NGFR reporter gene in the master cell line to the respective GPCR is 100% efficient, which is in accordance with previous studies. 11,12 In addition, the approach of the development of novel master cell lines ensured that the RMCE event occurs frequently. It was demonstrated that the number of correct targeting events is up to 10-fold higher than in previously established master cell lines. This allowed us to further reduce the time for RMCE-based cell line development down to 2 weeks, as described by Schucht et al. 11
For site-specific integration, other strategies also have been employed. Among them, for example, are the Flp-In system and Jump-In system (both from Invitrogen, Carlsbad, CA) and meganuclease-based approaches (cGPS system from Cellectis, Romainville, France); for review, see Wirth et al. 5 These technologies enable the site-specific integration of the desired gene expression cassette into the genome of a previously tagged host cell line (the respective master cell line). Recently, the Jump-In as well as the meganuclease approach has been successfully used for the establishment of HTS-compatible cell lines. 14,22
All these systems differ significantly from the RMCE approach presented here. In contrast to the cut-and-paste mechanism performed by the RMCE, the other systems (Flp-In, Jump-In, meganuclease) are best described as “paste” steps into chromosomal sites that were previously tagged by a first tagging vector. This pasting leads to the integration of two expression cassettes into the same genomic locus—the one used for the establishment of the master cell line (tagging cassette) and the one carrying the gene of interest. Such configurations of the expression units are prone to interferences (for review, see Gill et al. 23 ). For example, promoter interferences can arise when two transcription starts are in close proximity to each other. These promoter interferences can lead to an unpredicted expression to the point that the expression of the gene of interest is completely abolished. 11 Furthermore, this leads to the integration of vector backbone sequences that are CpG rich and thus have been shown to silence gene expression.
Indeed, it was shown that upon Flp-In-mediated transgene integration, the expression of the respective transgene was negatively influenced. 24 This report demonstrated that cell clones established with the Flp-In system display a high degree of variance despite their isogenicity. The authors found in isogenic clones different expression patterns ranging from homogeneous to mosaic or even silenced expression of the transgene.
The RMCE approach circumvents these problems as it is a molecular cut-and-paste mechanism. For the establishment of the master cell line, an expression cassette is employed that facilitates the tagging of a suitable genomic locus. In this study, a construct was used that expresses NGFR. This tagging cassette is completely removed when the desired expression construct is pasted in. Thereby, RMCE excludes phenomena as the mentioned promoter interference. Apart from this, the benefit of the approach described here is that the master cell line was specifically developed for the expression of transmembrane proteins. To enable robust screening campaigns or to facilitate purification of GPCRs (e.g., for crystallization studies), it is advantageous to use recombinant expression. Recombinant expression of GPCRs has been accomplished in all major expression systems ranging from Escherichia coli, yeast, and insect to mammalian cells. 25 In bacteria such as E. coli, overexpression of GPCRs often results in the accumulation of the receptor in inclusion bodies and is hampered by the lack of posttranslational modification. 26 Yeast provides a eukaryotic environment and ease of handling while still leading to high-level expression of the recombinant protein. However, mammalian cells are most likely to preserve structural and functional integrity when expressing mammalian proteins and are therefore the expression system of choice for drug development campaigns. Importantly, all recombinant cell lines presented here are stable in their expression characteristics and support drug screening efforts.
One major bottleneck in research is that GPCRs are difficult to express in mammalian cells. Strategies to solve this obstacle include optimization of codon usage 27–29 or inducible expression of the respective receptors. 28,30 Here it is shown that this limitation can be overcome by RMCE. With this approach, genomic loci are identified (and reused) that favor the expression of the recombinant gene.
The overexpression of transmembrane-spanning proteins such as GPCRs depends on a complex maturation process within the host cell, and this is known as a rate-limiting step to obtaining appropriate cell lines. 31,32 The correct folding and posttranslational modification leads to stress, which counterselects for nonexpressing cells. On the other hand, a few cells among a bulk of transduced cells are capable of performing high and stable expression. To specifically screen for those cells, the hypothesis was followed that expression is depending on the integration site or, in other words, that certain chromosomal regions support the expression of certain classes of proteins. To establish GPCR-expressing cell lines, we decided to use a receptor (NGFR) to screen for correct tagging sites. The results presented here demonstrate the validity of the hypothesis as the established GPCR-expressing cell lines did indeed show a robust and reliable expression pattern.
Beside GPCRs, other drug targets have become of high interest in the field of drug discovery—most important, ion channels and protein kinases. The current work and previous published approaches have shown that the RMCE strategy is flexible and can be adapted to various cells (of diverse origins) but also to various protein classes. Especially with respect to drug discovery, it is worth including the promising drug target classes of ion channels and kinases into RMCE-based cell line establishment efforts.
Furthermore, it has been shown that oligomeric proteins can be expressed from one targeted locus upon single-step integration. 11,33 Such an approach would suggest that also two different protein classes can be introduced at the same time—for example, the drug target and a specific reporter for monitoring the compound-induced target modulation.
In the field of GPCR research, other options are also imaginable. For example, to study heterodimerization, two different GPCRs can be integrated simultaneously into the same genomic locus, which should facilitate the expression of similar levels of both GPCRs. Alternatively, the desired GPCR could be potentially coupled to different arrestins or to different G-proteins to investigate the differential effects of these downstream effectors. In addition to the expression of heterologous proteins, the authors have used the RMCE strategy to integrate siRNAs to knock down the expression of cyclinD and reporter genes (unpublished data). The use of siRNAs can be used in this context to specifically knock down the expression of downstream factors of in/activated drug targets and to validate novel drug targets. Such approaches would maximize the potential of RMCE to become a flexible tool for the establishment of cell lines for drug discovery.
Footnotes
Acknowledgements
This work was supported by grants from the Helmholtz Society (Helmholtz Enterprise Fund), by the Niedersächsisches Ministerium für Wissenschaft und Kultur (80029155), and by grants of the German Ministry for Research and Education (FKZ 0313940 and PTJ-Bio/0313735/Nn07-06) and the Deutsche Forschungsgemeinschaft (Excellence Cluster REBIRTH).