
Abstract
Evaluation of drug cardiotoxicity is essential to the safe development of novel pharmaceuticals. Assessing a compound’s risk for prolongation of the surface electrocardiographic QT interval and hence risk for life-threatening arrhythmias is mandated before approval of nearly all new pharmaceuticals. QT prolongation has most commonly been associated with loss of current through hERG (human ether-a-go-go related gene) potassium ion channels due to direct block of the ion channel by drugs or occasionally by inhibition of the plasma membrane expression of the channel protein. To develop an efficient, reliable, and cost-effective hERG screening assay for detecting drug-mediated disruption of hERG membrane trafficking, the authors demonstrate the use of microfluidic-based systems to improve throughput and lower cost of current methods. They validate their microfluidics array platform in polystyrene (PS), cyclo-olefin polymer (COP), and polydimethylsiloxane (PDMS) microchannels for drug-induced disruption of hERG trafficking by culturing stably transfected HEK cells that overexpressed hERG (WT-hERG) and studying their morphology, proliferation rates, hERG protein expression, and response to drug treatment. Results show that WT-hERG cells readily proliferate in PS, COP, and PDMS microfluidic channels. The authors demonstrated that conventional Western blot analysis was possible using cell lysate extracted from a single microchannel. The Western blot analysis also provided important evidence that WT-hERG cells cultured in microchannels maintained regular (well plate-based) expression of hERG. The authors further show that experimental procedures can be streamlined by using direct in-channel immunofluorescence staining in conjunction with detection using an infrared scanner. Finally, treatment of WT-hERG cells with 5 different drugs suggests that PS (and COP) microchannels were more suitable than PDMS microchannels for drug screening applications, particularly for tests involving hydrophobic drug molecules.
Introduction
A
Currently, most hERG screening assays employ traditional well-plate formats and ultimately a GLP-compliant gold-standard patch-clamp electrophysiology approach to test for potential drug toxicity on model cell lines that stably express hERG ion channels. 11-13 Typical procedures involve selecting single cells for patch-clamp analysis with drug applied acutely to test for a reduction in hERG channel current 12 or mixing the drug of interest into cell culture plates for a desired time and then selecting single cells for patch-clamp analysis or lysates of cells for protein chemistry. Recently, live-cell Western analysis using infrared scanning technology was also employed to assess hERG surface membrane expression in a well-plate format. 14 Although a variety of new screening assays (e.g., automated patch clamp, radioligand displacement, flux assays, and HERG-Lite®15 ) have demonstrated increased throughput capabilities, there remains a need to further improve efficiency and lower cost of these assays. It has been estimated that the costs of drug development—from discovery to market—is between ~US$800 million and $2 billion per new drug, with a sizable portion of the costs consumed by synthesizing, storing, and testing numerous compounds that inevitably fail to reach market. 16-19 These costs can be lowered by reducing the volume of compounds necessary to perform large screening assays during the discovery stage of the development process. Microfluidics offers a low-volume alternative platform for testing drug toxicity of cultured cells. Microfluidic systems consist of arrays of microchannel geometries that can be designed for cell culture and tailored to model controlled microenvironments. Beyond the basic benefits of reduced reagent consumption and the potential for increased throughput, microfluidic systems allow precise spatial and temporal control of cell-cell and cell-matrix interactions. Because of the benefits of microfluidic cell culture, a number of studies in the past several years have investigated and characterized cell growth, maintenance, function, and response of various cell types in microfluidic devices. 20-29 However, to our knowledge, the properties of hERG-expressing cell lines in microfluidic cell culture have yet to be characterized. To prove that microfluidic channels are suitable for cell culture and analysis of hERG-expressing cells and to demonstrate the various advantages of microfluidic methods, it is necessary to properly characterize and validate the specific functional responses of these cells in microchannels.
In addition to characterizing the biological responses of hERG-expressing cells within microchannel environments, there is also a need to continue developing microfluidics technology that is more amenable to biological experimentation in the laboratory. An often-cited drawback of microfluidic systems, according to biologists, is their inherent complexity and need for external equipment, on-chip pumps and valves, and connecting tubes that are required for fluid handling. 30 Although some of these accessories can provide additional functionality and control within microchannels, they often lead to loss of efficiency and usability and to increased probability of technical failure (e.g., leaks). One technique that is both effective for handling flows in microchannels and amenable to biological experimentation is the use of surface tension-driven passive pumping. Passive pumping relies on dispensing precise volumes of droplets at the microchannel inlets and outlets and allowing differences in surface tension at droplet interfaces to produce a pressure gradient within the microchannel. 31,32 This method is convenient for biological assays because it only requires simple pipetting operations and can be integrated with automated liquid handling systems for high-throughput applications. 25 Furthermore, biological assays performed using microchannels can be analyzed using laser scanners and plate readers, as recently demonstrated, 23 and this further establishes microfluidics as a viable alternative for integration in the biology laboratory. Continued progress in this area is essential to the ongoing development of useful technology that can serve the biology research community.
Our objective in this study was to investigate the growth and culturing properties of human embryonic kidney (HEK) cells stably transfected with hERG channel protein toward the development of a high-throughput, reliable, and less costly platform for screening the cardiac toxicity of drug compounds. We cultured nontransfected and stably transfected HEK cells in polystyrene flasks, as well as in microchannels made from 2 different plastics (polystyrene [PS] and cyclo-olefin polymer [COP]) and a silicone elastomer (polydimethylsiloxane [PDMS]). We performed cell proliferation assays, immunofluorescence staining procedures, traditional Western blot analyses, and live-cell Western analyses coupled with infrared detection to establish microchannels as a viable culturing environment for hERG-expressing cells. In addition to the above-mentioned assays, electrophysiological patch-clamp experiments were performed on microchannel-cultured cells to verify the functional properties of the hERG ion channels after culturing in microchannels. After performing the necessary validation tests, hERG-expressing cells were further treated with various drugs to determine the suitability of microchannels for drug toxicity testing. Results demonstrated the potential for microfluidic screening and suggest that PS and COP (i.e., thermoplastic) microchannels may be more appropriate than PDMS devices because they do not undesirably absorb hydrophobic drug molecules. The use of passive pumping for fluid handling and infrared laser scanning for biological readout in our platform obviates the need for external pumps and tubing that are commonly used in other microfluidic systems and allows the use of existing biology laboratory infrastructure and equipment for analysis.
Materials and Methods
Microchannel devices
PS and COP devices were provided by Bellbrook Labs (Madison, WI) and consisted of a 16 × 12 array of straight microchannels each with 5.25 mm length, 1.0 mm width, and 140 µm height (surface area 8 mm2 and channel volume ~750 nL; Fig. 1A ). COP devices were used specifically for live-cell Western analyses of the 5 different drugs tested (see Drug Compounds below). PS devices were used for all other experiments conducted in plastic, including morphology experiments and fluoxetine treatment tests that initially compared PS to PDMS.
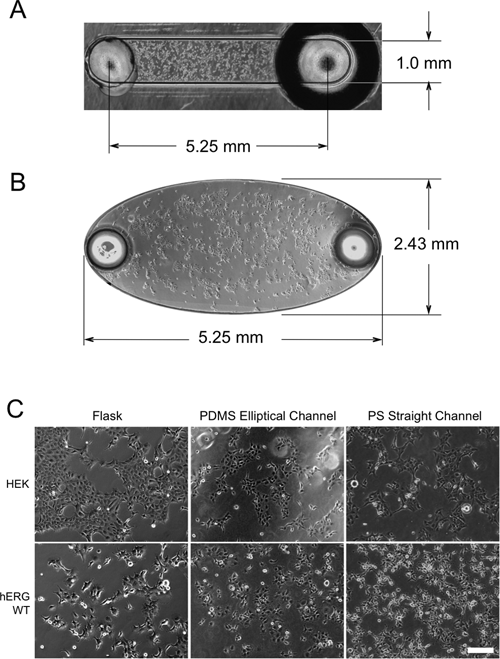
Culture of human embryonic kidney (HEK) and WT-hERG cells in (
PDMS elliptical microchannels (surface area 10 mm2 and channel volume 3.2 µL; Fig. 1B ) were fabricated using established soft lithography methods. 33 Briefly, SU-8-100 photoresist (Microchem, Newton, MA) was spin-coated onto a silicon wafer and soft-baked at 85°C for 4 h. The wafer was covered with a transparent photomask containing the desired pattern, exposed to UV light, baked after UV exposure at 85°C for 2 h, and developed in propylene glycol monomethyl ether acetate. After developing, the master was dried and hard baked at 85°C for 2 h.
PDMS casts were made by mixing prepolymer and curing agent (Sylgard 184 silicone elastomer kit, Dow Corning, Midland, MI) in a 10:1 ratio, then pouring the mixture onto the master and curing at 85°C for 4 h on a hot plate. After curing, the PDMS layer was peeled from the master and transferred to a clean glass dish.
Prior to cell culture, PDMS casts were autoclaved in a glass dish to sterilize the substrate and then passively bonded to flat PS plates (Omnitray, Nunc, Thermo Fisher Scientific, Rochester, NY). Microchannels were filled with serum-free minimum essential medium (MEM) and incubated overnight at 37°C. Microchannels were ready for cell culture once the medium was removed.
Cell culture
Normal (nontransfected) human embryonic kidney 293 (HEK) cells and transfected HEK cells that stably expressed wild-type hERG protein (WT-hERG) were used in the experiments. HEK cells were cultured in complete MEM (Gibco, Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS), 0.1 mM nonessential amino acids (NEAA), penicillin (100 U/mL) and streptomycin (100 µg/mL), and 1 mM sodium pyruvate and maintained at 37°C with 5% CO2. WT-hERG cells, previously described by Zhou and coworkers, 34,35 were cultured in the same complete MEM plus 400 µg/mL of the selective antibiotic geneticin (Gibco).
Culturing in microchannels
Prior to experiments, microchannels were incubated with culture medium for 30 min at 37°C. HEK and WT-hERG cells from culture plates were trypsinized, counted, and resuspended in culture medium at 1000 to 3000 cells/µL. Cells were seeded into microchannels via passive pumping by adding 2.0- or 3.2-µL droplets to the inlet port of PS/COP or PDMS microchannels, respectively. Microchannel devices containing cells were placed in a humidified tray covered with distilled water to prevent evaporation and incubated at 37°C with 5% CO2.
Cell proliferation assay
Cells were analyzed for proliferation by BrdU uptake. Cells were seeded at 300 or 450 cells/mm2 with medium containing 10 µM BrdU. At t = 24 h and 48 h, cells were fixed directly in the channels, and BrdU incorporation was detected by anti-BrdU staining. Briefly, cells were permeabilized with 0.2% Triton X-100 in phosphate-buffered saline (PBS), blocked with 3% bovine serum albumin (BSA) with 0.1% Tween-20 in PBS, and incubated with anti-BrdU antibody (Sigma-Aldrich, St. Louis, MO) overnight. Cells were then washed with PBS containing 0.1% Triton X-100 and incubated with Alexa Fluor 488 antimouse secondary antibody at 1:200 dilution. Images were taken using a CCD camera (SPOT RT monochrome, Diagnostic Instruments, Sterling Heights, MI).
TO-PRO-3 nuclear staining was also used to quantitatively measure cell proliferation. HEK and WT-hERG cells were seeded into microchannels at 300 cells/mm2 for PDMS microchannels and 450 cells/mm2 and 300 cells/mm2 for PS microchannels. Cells were fixed at 24 h and 48 h and then stained with TO-PRO-3 for 30 min after fixation. Nuclear staining signals were determined by imaging with an infrared scanner (LI-COR Odyssey; LI-COR, Lincoln, NE) using methods previously reported. 23
Immunofluorescence staining of hERG
Cells were seeded in PDMS microchannels on coverslips precoated with 1% (w/v) gelatin. Cells were fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.2% Triton X-100 in PBS for 10 to 15 min at room temperature. Cells were blocked with blocking buffer (3% BSA with 0.1% Tween-20 in PBS) for 1 h at room temperature and incubated with primary anti-hERG antibody (obtained from Craig January; 1:1000 dilution) overnight at 4°C. Cells were then washed 3 successive times for 5 min each with PBS containing 0.1% Triton X-100. Cells were incubated with Alexa Fluor 488 donkey antirabbit IgG antibody (Invitrogen) at 1:200 dilution for 1 h at room temperature. DAPI was added at 300 nM shortly before the end of the incubation. After washing cells for 3 times as above, the PDMS layer containing the microchannels was removed, and the coverslips were mounted on glass slides with mounting medium. Images were obtained by confocal microscopy (Bio-Rad Radiance 2100 MP; Bio-Rad, Hercules, CA).
Western blot analysis
Cells in microchannels were rinsed with PBS. Cell lysates were obtained by incubating cells with lysis buffer containing 1% Nonidet P-40, 150 mM NaCl, 50 mM Tris-HCl at pH 7.4, 5 mM EDTA, and 10% glycerol. Lysates were then collected from microchannels by careful aspiration. Protein concentration was determined using a micro-BCA protein assay reagent kit (Pierce, Thermo Scientific). Equal amounts of protein were subjected to electrophoresis on a 4% to 20% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel (Invitrogen) followed by electrophoretic transfer onto nitrocellulose membranes. After transfer, the nitrocellulose membranes were blocked for 1 h with blocking buffer (LI-COR) containing 0.1% Tween-20 and then incubated with rabbit anti-hERG polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA; 1:1000 dilution) and antiactin monoclonal antibody (MP Biomedicals, Solon, OH; 1:10,000 dilution) at room temperature overnight. The protein bands were visualized with IR Dye 800 CW–conjugated donkey antirabbit IgG (Rockland, Gilbertsville, PA) and IR Dye 680–conjugated goat antimouse IgG (LI-COR) antibodies both at 1:10,000 dilution.
Drug compounds
Five different drugs with known hERG-related toxicity levels were used to test the microchannel platform. Fluoxetine was used as a model drug for testing drug effects on cell morphology (via phase contrast microscopy) and on hERG protein expression (via both live-cell Western and conventional Western blotting). Four other drugs were used to further validate the robustness of the live-cell Western analysis using the infrared scanner. Aspirin (ASA) and acetaminophen (ACE) were chosen as common drugs known to have no hERG-related toxicity. Ivermectin and arsenic trioxide (As2O3) were chosen for their known effect of inhibiting hERG trafficking to the cell surface, which is in contrast to the dual effects of fluoxetine (i.e., both direct hERG blocking and inhibition of hERG trafficking). 15 All 5 drugs were acquired from Sigma-Aldrich. ASA and ACE were dissolved in ethanol, ivermectin was dissolved in DMSO, and fluoxetine and As2O3 were dissolved in deionized water, as specified.
Live-cell Western of hERG
Cells were seeded into 1% (w/v) gelatin-coated PDMS elliptical microchannels or PS and COP straight microchannels and cultured overnight. After overnight culture, WT-hERG cells were treated with the 5 different drug compounds at desired concentrations. After 22 h, cells were incubated with primary anti-hERG antibody (Alomone Labs, Jerusalem, Israel; 1:50 dilution) for 2 h at 37°C. This antibody binds to the extracellular S1-S2 linker domain of the functional 155-kDa hERG protein of live cells, permitting live-cell detection of protein expression. Cells were washed 3 successive times for 10 min each with complete cell culture media. Cells were then incubated with IR Dye 800 CW–conjugated donkey antirabbit IgG antibody (Rockland) at 1:500 dilution in complete cell culture medium for 1 h, followed by incubation with Syto 60 (Invitrogen) at 1:10,000 dilution for 30 min. Cells were washed 2 more times for 10 min each with complete cell culture media followed by 2 washes with PBS. Cells were imaged with an infrared scanner (LI-COR Odyssey) using methods previously reported for live-cell Western analyses. 14,23
Electrophysiology
Cells were seeded in PS channels as described above for live-cell Western analysis. To prepare for patch-clamp experiments, cells were incubated with 0.25% trypsin-EDTA for 2 min after brief washing with PBS. The cell suspension was then pipetted into a 0.5-mL tube containing 100 µL of complete medium. Cells were centrifuged at 150 g, and the supernatant was removed by aspiration.
hERG current (I hERG) was measured using the whole-cell patch-clamp technique as described previously. 14,34,35 The extracellular (bath) solution contained 137 mM NaCl, 4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH 7.4 with NaOH), and the intracellular pipette solution contained 130 mM KCl, 1 mM MgCl2, 5 mM EGTA, 5 mM MgATP, and 10 mM HEPES (pH 7.2 with KOH). The holding potential was −80 mV. The cells were depolarized to 50 mV for 4 s to maximally activate hERG channels, followed by a test pulse to −50 mV for 5 s to measure the peak tail I hERG. All voltage clamp experiments were performed at 22 to 23°C within 1 to 2 h after removing the cells from their culture conditions. Voltage protocols and data analyses were done using the pCLAMP 8.0 (Axon Instruments, Union City, CA) and Origin (6.0 and 7.5, Microcal, North Hampton, MA) computer software.
Results
HEK and WT-hERG (transfected HEK) cells were seeded and cultured in PS straight microchannels, COP straight microchannels, and PDMS elliptical microchannels and compared to cells in PS flasks. Cultured cells were evenly distributed in both microchannel geometries (
Proliferation of HEK and WT-hERG cells in microchannels was examined by measuring BrdU incorporation directly in the microchannels. Anti-BrdU staining showed that both HEK and WT-hERG cells cultured in PS microchannels were actively incorporating BrdU at 24 h (
Fig. 2A
) and 48 h (data not shown). To examine overall growth rate of cells in microchannel culture, cell proliferation was also assessed by nuclear staining using TO-PRO-3 dye. Previous results from our lab demonstrated that TO-PRO-3 intensity varies linearly with cell number.
23,25
In the current study, TO-PRO-3 staining intensity (and thus cell count) increased significantly from 24 to 48 h (
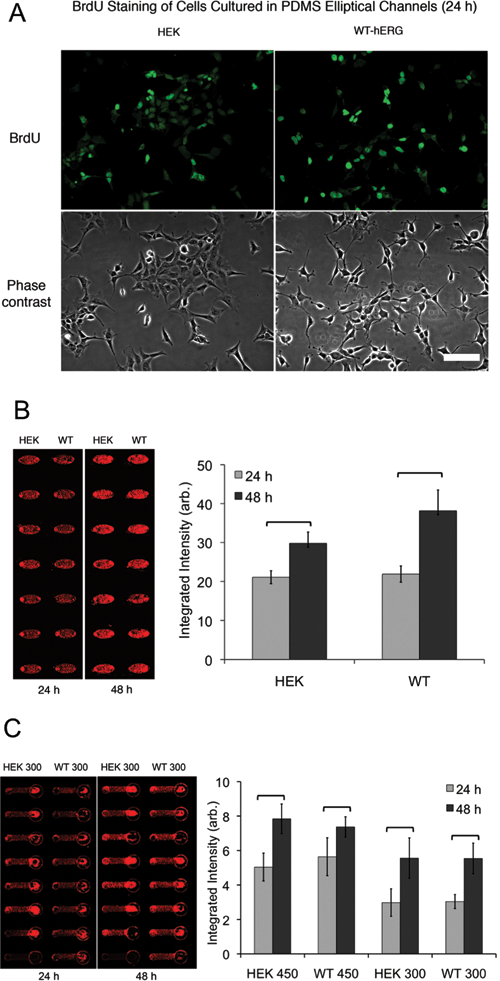
Human embryonic kidney (HEK) and WT-hERG cell proliferation was determined by (
In addition to measuring proliferation by nuclear staining, immunofluorescent staining of hERG protein was also performed to determine the localization of hERG ion channels in WT-hERG cells. Cells showed distinct membrane and intracellular staining of hERG protein ( Fig. 3A ), similar to published images of cells cultured in conventional PS flasks. 34,35 To further characterize WT-hERG cells, hERG protein expression was determined by Western blot analysis ( Fig. 3B ). Culture of WT-hERG cells results in 2 types of hERG protein: the functional, N-linked glycosylated mature form located on cell membranes (155 kDa) and the immature core-glycosylated form located in endoplasmic reticulum (135 kDa), and these are shown in the Western blot. Although cell counts are low in microchannels, and therefore low amounts of cellular protein product may make Western blotting challenging from microchannel culture, we were able to detect hERG expression from a single elliptical PDMS microchannel by using sensitive infrared dyes together with a stably expressed hERG cell line. As shown in Figure 3B , WT-hERG cells readily expressed both forms of hERG protein to confirm that WT-hERG cells cultured in microchannels maintained biochemical properties. To confirm that the expressed hERG channels maintained biological function, patch-clamp experiments were performed on WT-hERG cells cultured in microchannels. These experiments demonstrated that the cells maintained large-amplitude currents with normal biophysical properties ( Fig. 3C ).
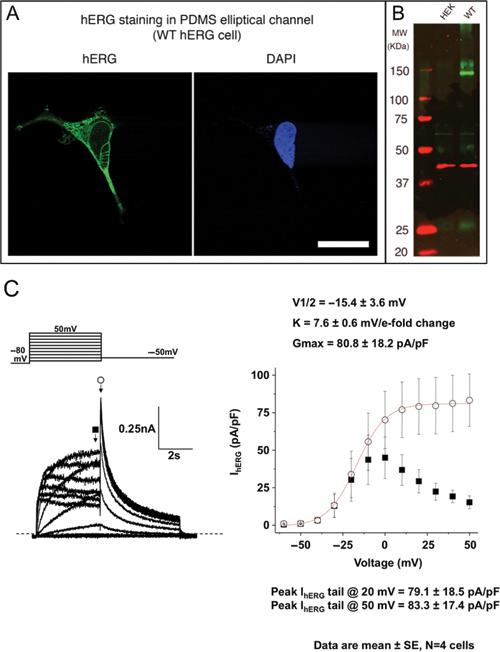
Characterization of hERG expression and K+ channel function after microchannel culture. (
After characterizing hERG protein expression and function from WT-hERG cells cultured in microchannels, we further investigated the effects of drug treatment on WT-hERG cells. We first used fluoxetine (Prozac), an antidepressant drug that at higher concentrations directly blocks hERG ion channels as well as disrupts its protein trafficking. 37 Fluoxetine at 30 µM and 60 µM was added to microchannel-cultured WT-hERG cells. In 96-well plates, both 30 and 60 µM of fluoxetine visibly lowered cell viability. In contrast, although 60 µM dose of fluoxetine noticeably decreased cell viability in PS and COP microchannels, 30 µM did not appear to affect cell viability or morphology ( Fig. 4A for PS microchannels). Live-cell Western analysis was used to test the effect of fluoxetine on hERG channel protein expression in WT-hERG cells cultured in 96-well plates and in PDMS, PS, and COP microchannels. In previous reports, fluoxetine treatment was shown to decrease the expression of the mature form of hERG because of abnormal trafficking of the hERG channel protein to the surface membrane. 37 We performed live-cell Western analysis of WT-hERG cells in 96-well plates and showed that, as expected, WT-hERG cells had positive surface expression of functional hERG protein, whereas HEK cells did not express hERG protein. More important, surface expression in WT-hERG cells was reduced after 22 h of 30 µM fluoxetine treatment, confirming previous reports. 37 Similarly, live-cell Western analyses were performed in the different types of microchannels. In PS and COP microchannels, hERG protein expression was clearly affected by increasing concentrations of fluoxetine treatment. Statistically significant differences (p < 0.01) were found between 0-, 30-, and 60-µM treatment conditions on WT-hERG cells in PS microchannels ( Fig. 4B ). These findings were similar to those in COP microchannels, and the findings also exhibited a similar trend to results from 96-well plates after fluoxetine treatment. In contrast to the results from 96-well plates and PS microchannels, however, WT-hERG cells cultured in PDMS microchannels did not respond to fluoxetine even when drug concentration was increased to 480 µM, an 8-fold increase from PS microchannel tests (discussed below). To further verify the live-cell Western results for hERG protein expression in microchannels, standard Western blot analysis was performed with cells cultured in PS microchannels. Results showed that the expression of the functional 155-kDa form of hERG protein was reduced after fluoxetine treatment, confirming live-cell Western results from microchannels ( Fig. 5 ).
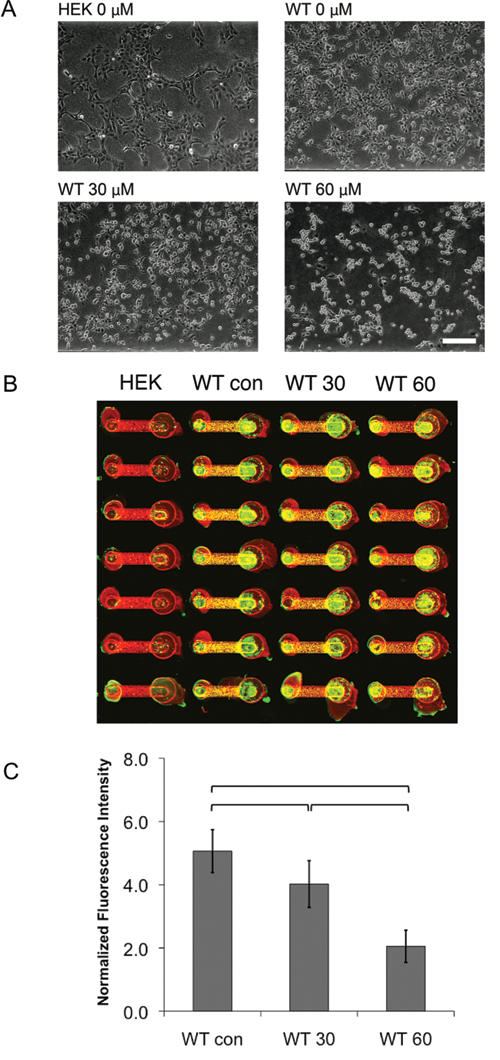
Fluoxetine treatment of WT-hERG cells in polystyrene (PS) microchannels. (
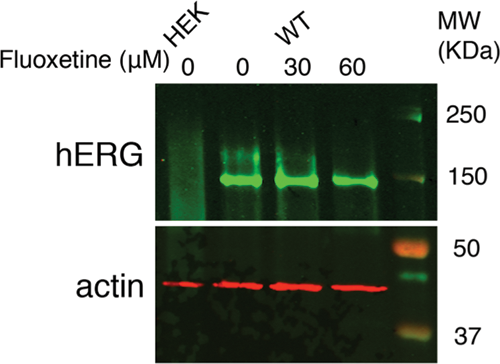
Conventional Western blot analysis of hERG protein expression after fluoxetine treatment of human embryonic kidney (HEK) and WT-hERG cells cultured in polystyrene microchannels.
In addition to using fluoxetine as a model drug for detailed analysis of hERG protein expression of cells cultured in microchannels, we also tested a small panel of drug compounds to validate the robustness of the live-cell Western method in a microfluidic platform. To cover a range of drug responses, we tested 4 other drugs with well-documented effects on hERG. ASA and ACE were used as examples of drugs with no effect on hERG, whereas arsenic trioxide (As2O3) and ivermectin (Iver) were chosen as drugs that would induce hERG-related toxicity via inhibition of hERG ion channel trafficking to the cell surface membrane. As expected, both ASA and ACE, at concentrations as high as 60 µM, showed no significant difference of hERG staining intensity compared to control conditions (i.e., both wild-type [WT] control and ethanol [p > 0.1]; Fig. 6 ). In contrast, As2O3 at 10, 30, and 60 µM all showed significant decreases of hERG staining intensity compared to no treatment (p < 0.001). Ivermectin at 30 µM showed a significant decrease of hERG staining intensity compared to both no treatment and DMSO controls (p < 0.001), but ivermectin at 10 µM did not show any significant differences to controls (p > 0.1). All results were consistent with other reports of these drugs on hERG-related toxicity, except for the case of ivermectin at 10 µM. 15 Similar to the results of fluoxetine at 30 µM, ivermectin at 10 µM did not produce the same toxic effects in microchannels as it produced at the macroscale, suggesting that higher concentrations of drug are necessary in microchannels to induce the same macroscale effect.
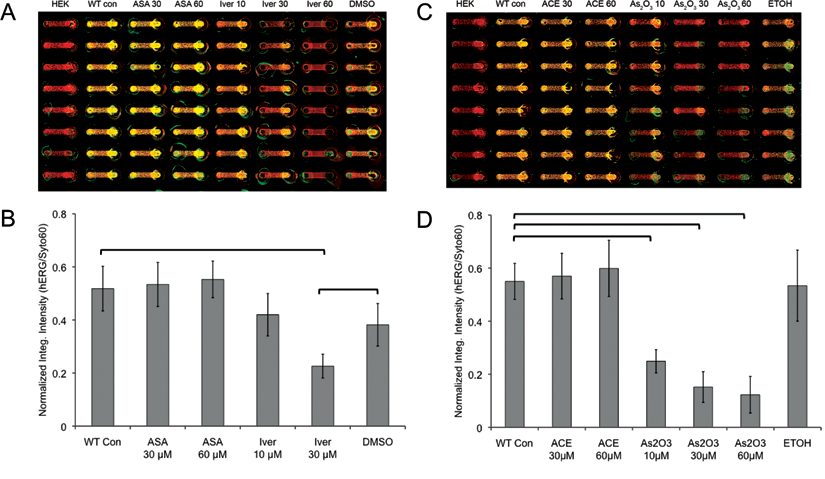
Live-cell Western analyses for various drug treatments of WT-hERG cells in cyclo-olefin polymer (COP) microchannels. (
Discussion
The hERG K+ ion channel is important in mediating cardiotoxicity of many drug compounds. 3 Block of hERG channels reduces the key repolarizing current, I Kr, to cause QT interval lengthening on electrocardiogram (EKG) to potentially induce the life-threatening cardiac arrhythmia torsades de pointes. Cell lines that stably express hERG have been developed to facilitate their use as a screen for drug-induced QT prolongation. 34,35 Despite major research efforts to develop reliable, affordable hERG screens, existing hERG screening assays are largely inefficient and expensive because they require relatively large volumes of reagents, a large number of lead compounds, and considerable manual labor. Microfluidics has promised to provide a feasible alternative to conventional drug screening platforms by lowering reagent consumption, improving sensitivity and convenience of detection methods, and increasing throughput. To show that microfluidics can be used to facilitate the development of a high-throughput drug screen, we cultured HEK and—for the first time—WT-hERG cell lines in microfluidic channels and characterized cell behavior and hERG protein expression of cells cultured in microchannels. Results demonstrated that microfluidic channels have the potential to improve current cardiotoxicity drug screening methods.
The current study demonstrated a number of the aforementioned advantages of using microfluidics for cell-based drug screening and other applications. Large arrays of microchannels coupled with direct immunostaining and infrared scanning methods can streamline experimentation, increase efficiency, and improve quality of cell imaging by avoiding meniscus effects that adversely affect image quality in 96- and 384-well plate formats. The low volumes in microchannels (less than 10 µL) help to reduce reagent consumption considerably (50-fold decrease in volume compared to 96-well plate) 36 but are particularly advantageous for investigations that involve limited cell numbers from rare cell populations and precious patient samples. Specifically for drug screens, such low-volume requirements essentially mean that much smaller quantities of early stage compounds need to be synthesized, resulting in significant savings during early trials. We were able to demonstrate that the sensitivity of infrared dyes for direct in-channel immunostaining was sufficient for quantitative analyses of cell responses even for low cell numbers in microchannels. Furthermore, we were able to demonstrate for the first time that cell lysate from a single microchannel (O(~104) cells)) is sufficient for detection of the hERG protein, an important step in establishing the feasibility of using microfluidics for cardiotoxicity drug screening.
Although many other microfluidic systems similarly offer increased efficiency and reduced reagent consumption, these other systems typically rely on external pumps and tubing to introduce reagents into the microchannels. 38 Our platform is distinct from these other systems because we employ passive pumping of droplets to perform fluidic volume replacements, thus eliminating the need for external pumps and tubing. Because of the use of passive pumping, cell loading and reagent replacement can be conveniently performed by pipetting droplets to microchannel ports, and automation with liquid handling systems can be potentially employed for large-scale operations. 25,39 Indeed, it is conceivable to adapt our passive pumping methodology to automate a nonradioactive rubidium (Rb) efflux assay as a metric for hERG channel function. 40 Rb efflux assays are becoming increasingly popular for functional tests of hERG, and our passive pumping-based microchannel platform facilitates cell culture and subsequent collection of conditioned media and cell lysates for off-chip analysis. Notably, hERG radioligand displacement assays offer the same potential. 41
Since microfluidics offers clear advantages over existing platforms, it is an important first step to validate its appropriateness specifically for hERG screening. To verify the suitability of microfluidic channels for hERG screening, HEK and WT-hERG cells were cultured in regular PS flasks, straight PS microchannels, straight COP microchannels, and elliptical PDMS microchannels. PS and PDMS microchannels were tested because these 2 materials represented the most widely used materials for tissue culture plasticware and microchannel devices, respectively; COP has become increasingly popular as a material for biology experiments, as evidenced by recent advances in commercially available assay technologies (e.g., Bellbrook Labs). It was important to determine which material was most suitable for our application. Although 2 different geometries were used (straight vs elliptical), the geometry was not expected to cause differences in culturing results because the surface area-to-volume ratios were of the same order of magnitude (i.e., O(10−2) m−1). PS and COP microchannels were straight because they were designed by Bellbrook Labs for general applications that would appeal to a large commercial market. PDMS microchannels, on the other hand, were elliptical because the shape was easily fabricated using soft lithography (no more difficult than fabrication of straight microchannels), and it provided uniform distribution of cells during seeding as well as a larger cell culture area for higher fluorescence intensity measurements. In both types of microchannels, HEK and WT-hERG cells were cultured successfully, with cells exhibiting normal morphology and proliferation rates, typical protein expression of hERG as determined by Western blotting, and normal electrophysiological properties as determined by standard patch-clamp experiments. These experiments validated WT-hERG cells as an appropriate model cell line for microfluidic cardiotoxicity drug screening. The results also confirmed that geometry did not affect the culturing of HEK and WT-hERG cells and suggested that PS, COP, and PDMS were suitable materials for a screening platform. Subsequent experiments with a model drug, fluoxetine, revealed an important difference between PS/COP and PDMS devices. Using live-cell Western analyses of hERG expression in WT-hERG cells treated with fluoxetine in thermoplastic and PDMS microchannels, it was discovered that WT-hERG cells responded to fluoxetine in thermoplastic microchannels but not in PDMS microchannels. The absence of any measureable drug effects in experiments using PDMS microchannels was likely due to the ability of PDMS to absorb and sequester fluoxetine, a hydrophobic molecule, into the bulk material. Work in our lab has shown that PDMS possesses undesirable characteristics for culturing cells—namely, its absorption of small hydrophobic molecules and its release of uncrosslinked PDMS oligomers into the channel fluid. 42,43 Although these issues are less concerning when nonhydrophobic molecules are used in drug treatment 44 or when solvent extraction is used to remove most of the uncrosslinked PDMS oligomers, these drawbacks limit the potential acceptance of PDMS as a material for robust biological applications. The results from our drug treatment tests in PS and COP microchannels, however, showed expected response of WT-hERG cells that were similar to results in PS flasks. This suggests that microchannels made of PS or COP may be more appropriate for drug screens, particularly since PS and COP are more mainstream than PDMS in lab cultureware, and neither thermoplastic has issues related to the absorption of small hydrophobic molecules.
Although PS and COP microchannels performed as expected for most of the results, one interesting finding was that both fluoxetine and ivermectin needed higher concentrations in microchannels to induce the same toxic effects as seen in well plates. This difference was likely due to the large increase in surface area-to-volume ratio in microchannels compared to wells, resulting in a proportionate decrease in drug per unit area of culture surface or, ultimately, in drug molecules per cell. Thus, drug concentrations may need to be adjusted accordingly for microchannel experiments based on the known change in surface area-to-volume ratio from macroscale well experiments.
Conclusions
We successfully demonstrated microchannel cell culture, drug treatment, protein analysis, and immunostaining of normal HEK cells and transfected HEK cells stably expressing hERG. Our results provided the necessary groundwork for pursuing a microfluidic-based drug screening assay for hERG-related cardiotoxicity that has the potential to improve efficiency and lower costs while opening the door to potential improvements in spatial and temporal control of the cellular microenvironment. The assay is potentially useful for screening for many other forms of acute and subacute toxicity in many cell types. Furthermore, our techniques are readily amenable to the development of other cell-based assays for drug development, including assays for ADME screening, and reporter-based assays for gene regulation studies. Specifically, our results indicated that PS, COP, and PDMS are in many ways comparable materials for microchannel cell culture, but the differences that do exist between them can lead to distinct biological outcomes when drugs and other soluble factors are involved in the experimental study. In such sensitive microenvironments as cell culture within microchannels, PS and COP demonstrate the necessary inertness to biochemical species that will allow their future use in microscale cell culture and drug screen applications.
Footnotes
Acknowledgements
D. J. Beebe has an ownership interest in BellBrook Labs, LLC, which has licensed technology presented in this manuscript. Dr. T. Kamp and C. January are cofounders of Cellular Dynamics International. We acknowledge financial support from NIH grants R21CA122672, K25CA104162, and R01 HL60723. We also acknowledge assistance from Keil J. Regehr for microchannel fabrication and Katherine M. Holzem for help with hERG Western blotting.