
Abstract
Human-induced pluripotent stem cells (HiPSCs), and new technologies to culture them into functional cell types and tissues, are now aiding drug discovery. Patient-derived HiPSCs can provide disease models that are more clinically relevant and so more predictive than the currently available animal-derived or tumor cell-derived cells. These cells, consequently, exhibit disease phenotypes close to the human pathology, particularly when cultured under conditions that allow them to recapitulate the tissue architecture in three-dimensional (3D) systems. A key feature of HiPSCs is that they can be cultured under conditions that favor formation of multicellular spheroids or organoids. By culturing and differentiating in systems mimicking the human tissue in vivo, the HiPSC microenvironment further reflects patient in vivo physiology, pathophysiology, and ultimately pharmacological responsiveness. We assess the rationale for using HiPSCs in several phases of preclinical drug discovery, specifically in disease modeling, target identification, and lead optimization. We also discuss the growing use of HiPSCs in compound lead optimization, particularly in profiling compounds for their potential metabolic liability and off-target toxicities. Collectively, we contend that both approaches, HiPSCs and 3D cell culture, when used in concert, have exciting potential for the development of novel medicines.
Introduction
For decades, drug discovery has employed automated liquid handling and sensitive detection instrumentation for use in high-throughput screening (HTS) systems to identify novel compounds as leads to treat human diseases.1–8 The widespread use of cellular assays, in both target-based and phenotypic compound screening, is now standard practice.2,6 In general, the cells used in HTS campaigns have been derived from either human tumor cell lines (usually transformed to overexpress the desired molecular targets) or animals, frequently rodents.2,9,10 Human primary cells are used infrequently, due to issues of abundance and reproducibility. 2 However, the development of technologies to generate large and reproducible numbers of human-induced pluripotent stem cells (HiPSCs) has led to the use of HiPSCs as a source of primary human cells for disease modeling, and in high-throughput screens for lead identification, validation, and optimization.
HiPSCs provide advantages for drug discovery over the use of rodent or tumor cell lines.1,2,11–14 First, the cells are derived from the species (human) for which the test compounds are intended to be used therapeutically. Second, technologies are now available to culture HiPSCs (with the desired genetic profile) to provide a virtually unlimited, yet reproducible, supply of human cells for HTS of large compound libraries. Third, the patient-derived HiPSCs can be differentiated to specific cell types endogenously expressing the molecular target of interest, as well as other tissues and organs, for example, liver, muscle, kidney, heart, and central or peripheral nervous system neurons, to test for off-target effects of drugs under development.
The key advantage of HiPSCs lies in their ability to be differentiated to specific human cell types of interest, to allow for the selection of compounds that preferentially modulate the activity of those cells with limited activity at other cell types. HiPSC-derived neurons, for example, can be used to screen for compounds that affect activity at central nervous system (CNS) targets, including compounds that may modulate neuronal transmission.4,5,11–14 Active compounds identified can then be profiled for their selectivity, as judged by their lack of activity on other human tissues, including hepatocytes, cardiac myocytes, skeletal muscle, or gastrointestinal tissue.
Several approaches exist to differentiate HiPSCs into relatively pure cell line populations in the requisite abundance and reproducible amounts required for HTS campaigns.1,15 Procedures have also been developed to co-culture cells from the same patient.16–20 HiPSCs, for example, can be differentiated into motor neurons, which then can be co-cultured with skeletal muscle cells from the same individual.16–19 These co-cultured cells can form active neuromuscular junctions. Since neuromuscular junctions are key sites in the body to control movement, these organoids can be used to screen for compounds that improve motor function. By this means, novel agents may be identified that can be developed to treat patients with debilitating movement disorders, such as amyotrophic lateral sclerosis (ALS) or traumatic spinal or muscle injuries, or elderly patients in whom motor control has declined with age. Similarly, HiPSC co-culturing protocols have been used to generate human intestinal tissue organoids with a functional enteric nervous system. 21 These organoids can be used to develop a new generation of drugs to treat gastrointestinal disorders.
As mentioned above, HiPSCs are now emerging as powerful tools to develop models of human disease ( Table 1 ).11–13 Specifically, models for neurodegenerative diseases, including Alzheimer’s disease (AD),22–24 ALS,25–28 frontotemporal dementia (FTD),29–32 Huntington’s disease (HD),33,34 and Parkinson’s disease (PD),35–39 have been developed, all of which employ HiPSC lines derived from patients with familial forms of the disease. These disease models express the patient’s genetic background and exhibit disease phenotypes, including accelerated neurodegeneration, and allow both single- and multi-time-point studies (sometimes referred to as “4D” culture) to be conducted. Furthermore, patient-derived HiPSCs can be directed to differentiate into neuronal subpopulations that are most vulnerable in each disease, for example, motor neurons (ALS), dopamine (DA) neurons (PD), and striatal neurons (HD). Other cell culturing protocols have been developed that enable the generation of neuronal populations directly from patient fibroblasts.40–43 In this case, the cells maintain both the patient’s epigenetic markers and the age characteristics, thereby providing important models for investigating sporadic forms of the neurodegenerative diseases.
Human iPSC Models of Neurodegenerative Diseases.
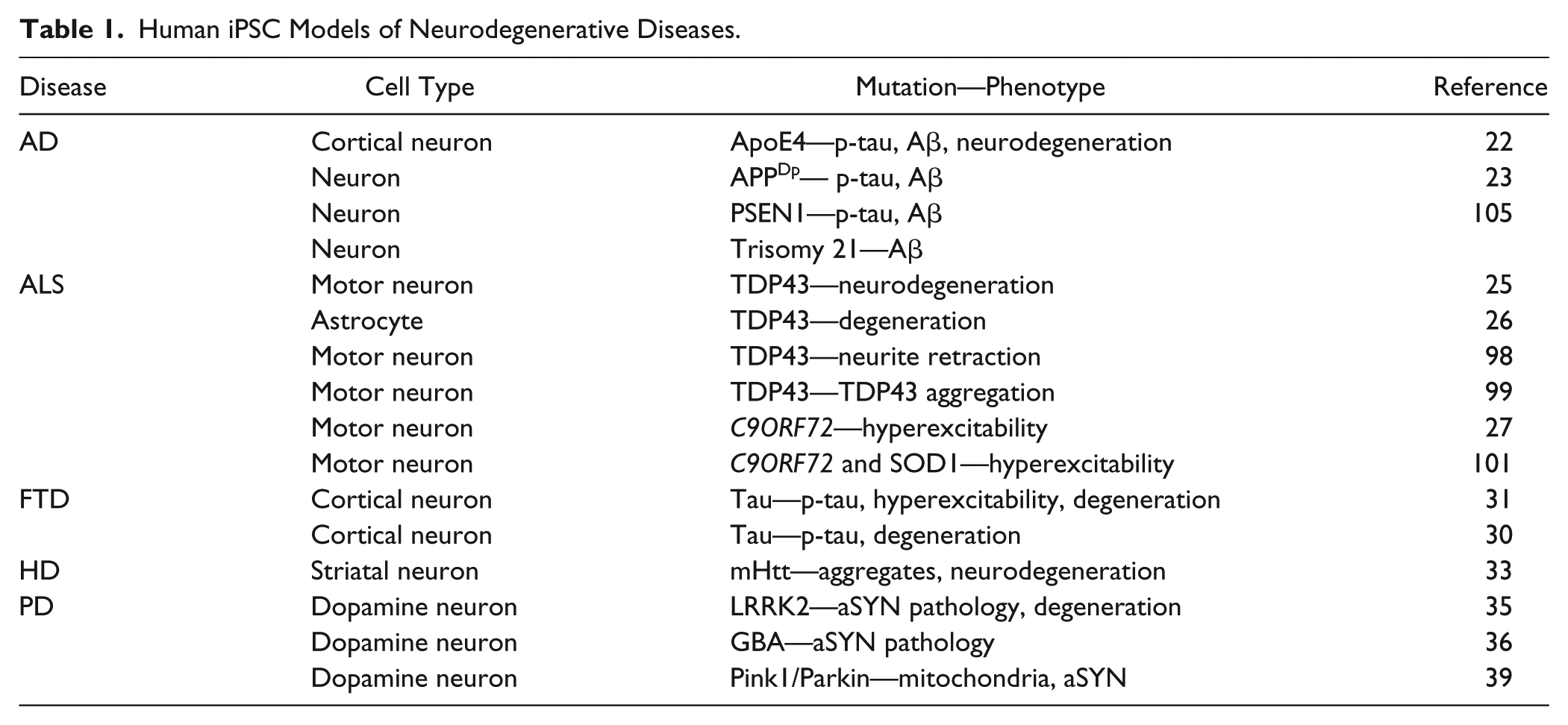
The use of HiPSCs from different patients (but with the same disease) can also facilitate understanding of the influence of individual variation in compound pharmacology, prior to clinical evaluation. Taking HiPSC-derived neurons as illustrative of this emerging field, one can envision in the future an increase in the use of HiPSCs—derived from large patient cohorts—for both disease modeling and compound screening for efficacy and safety; these data could be correlated (i.e., “translated” from preclinical to clinical study) with individual patients’ clinical histories, allowing precise patient stratification during clinical evaluation.
Human iPSCs Used for Drug Discovery and Safety Evaluation
Frequently, HiPSCs are isolated from skin fibroblasts and then generically “reprogrammed” to an embryonic-like state by viral introduction of transcription factors that induce pluripotency.1,44 The resulting iPSCs are self-renewable but can also be directed to differentiate into different cell types. With specific reference to those cells needed for preclinical lead optimization, HiPSCs can be differentiated into liver hepatocytes, cardiac myocytes, skeletal muscle, gastrointestinal cells, and neurons, by ectopic expression of genetic drivers, growth factors, and/or small molecules. 1 During this process, HiPSCs can also be genetically modified to express biosensors, usually fluorescent or luminescent probes, to provide simple biochemical or imaging HTS assays to assess cell morphology, function, and survival.11–14
Human Liver Cells
HiPSC-Derived Hepatocytes for Toxicity Evaluation
Primary human hepatocytes can be used to profile compounds for their potential to induce liver toxicity and for their metabolic liability in patients. Because human hepatocytes may respond differently to drugs than rodent- or tumor-derived hepatocytes, their use provides major advantages in testing for drug safety at the preclinical discovery stage. However, the source of these cells is limited, restricting their widespread use in drug safety screens.
By contrast, human hepatocytes derived from HiPSCs can be produced in high abundance and reproducibility, with the desired genetic profile. Sirenko et al.45,46 and others51,52 have reported the use of HiPSC-derived hepatocytes in preclinical profiling programs, showing that toxicity could be assessed by imaging phenotypes reflective of the cell’s health, including morphology, adhesion and spreading, nuclear condensation, cytoskeleton integrity, and mitochondria membrane potential. Compound screening of potentially hepatotoxic compounds against HiPSC-derived hepatocytes yielded similar results as when the same compounds were tested against primary human hepatocytes. These data and others validated the use of HiPSC-derived hepatocytes for preclinical lead optimization, and showed that the cells had a high degree of clinical predictability.
HiPSC-Derived Hepatocytes for Compound Metabolism Profiling
HiPSC-derived hepatocytes can also be used to evaluate the metabolic liability of compounds and have the potential to be used to assess the influence of patient genetic variability on drug metabolism. 47 Specifically, genetic variability may cause differences from one individual to another in P450 enzyme expression, which can result in individuals being either “slow” or “fast” metabolizers of many commonly prescribed drugs. 48 The utility of the HiPSC-derived hepatocytes in this regard was reported by Takayama et al., 47 who used HiPSC-derived hepatocytes isolated from individuals with or without a single-nucleotide polymorphism (SNP) in the P450 enzyme, CYP2D6; the SNP in the CYP2D6 genes reduces enzymatic activity.48,50 Consequently, individuals with this SNP are slow metabolizers of drugs, including tamoxifen. Takayama et al. 47 then cultured MCF7 human breast cancer cells with HiPSC-derived hepatocytes from individuals with or without the CYP2D6 SNP. Normally, CYP2D6 metabolizes tamoxifen to a cytotoxic metabolite in breast cancer cells. 49 Co-cultures of cancer cells with hepatocytes derived from individuals with the normal CYP2D6 enzyme showed substantial loss of cancer cells after tamoxifen treatment. As expected, cancer cells co-cultured with hepatocytes from individuals with the CYP2D6 SNP showed resistance to tamoxifen, because less of the cytotoxic metabolite was produced, and ultimately diminished breast cancer cell toxicity. These data support the potential of HiPSC-derived hepatocytes in evaluating the influence of CYP polymorphisms on drug toxicity and metabolism. This may be particularly important when using human cell culture protocols to generate hepatocytes directly from fibroblasts, since these hepatocytes would be predicted to better retain the epigenetic as well as genetic profile of the human donor.
Humans vary in the expression levels of P450 enzymes, and therefore metabolize drugs differently. 53 HiPSC-derived hepatocytes therefore provide better models of the diversity of metabolic responses existing in patient populations. Therefore, they may provide better predictors of clinical efficacy and safety than many other models of liver function.
These properties of HiPSC-derived hepatocytes may also facilitate the development of unique multi-tissue/organ systems to determine human drug toxicity. 54 End-organ toxicity is a major limiting factor in drug development. Consequently, approaches that predict preclinical drug safety may provide the means to reduce the attrition rate of novel leads in the clinic. In this respect, microfluidic systems have proven to be very useful. Here, different organoids derived from HiPSCs—such as hepatocytes, cardiac myocytes, gastrointestinal and kidney cells, or neurons—are grown in separate, but interconnected, chambers, and can be used to profile drugs and their metabolites simultaneously for potential toxicity in different tissues in the body. 55
HiPSC-Derived Hepatocytes for Drug Discovery
HiPSC-derived human hepatocytes can also be employed in drug discovery against molecular targets that cause liver disease. 56 For example, HiPSC-derived hepatocytes generated from patients with homozygous familial hypercholesterolemia have provided an approach to identify compounds that lower serum low-density lipoprotein C (LDL-C). Cayo et al. 56 have identified several cardiac glycosides that lowered expression of apolipoprotein B in the human hepatocytes in vitro. In accordance with these data, patients treated with these same cardiac glycosides also had reduced serum LDL-C levels, collectively supporting the potential use of cardiac glycosides to treat hypercholesterolemia.
The use of HiPSC-derived hepatocytes may also provide unique models to study infectious diseases, including hepatitis B (HBV) and hepatitis C (HCV).57–60 In this case, animal models are suboptimal in studying HBV and HCV, primarily because the viruses infect humans. Thus, animal- or tumor-derived hepatocytes provide limited utility to study the infectivity of these viruses.
Several groups have now demonstrated that HiPSC-derived hepatocytes express HCV and HBV entry receptors and support productive viral infection with HCV or HBV particles.58,61,62 The viral load in HiPSC-derived hepatocytes is, in fact, higher than that in hepatoma cell lines. Furthermore, the antiviral innate immune response to HCV or HBV infection in the HiPSC-derived hepatocytes can be detected, with increased expression of various type I interferon-stimulated genes, like that observed in primary human hepatocytes. These findings, taken together, suggest that as HiPSC-derived hepatocytes predictably respond to viral infection, they can be used to identify new, more effective, and safer human anti-HBV and HCV anti-infective agents.
Human Cardiac Myocytes
HiPSC-Derived Cardiac Myocytes to Toxicity Evaluation
Several cell culture technologies are now available to generate HiPSC-derived cardiac myocytes for use in the evaluation of drug toxicity. 63 Indeed, drug-induced cardiac toxicity is a major risk in therapeutic development;55,63–68 the U.S. Department of Health and Human Services estimates that nearly 1 million patients suffer from adverse drug reactions each year, with drug-induced arrhythmia being the leading cause. 71 Notably, it is the potential of novel drugs to induce prolongation of the QT interval that leads to sudden cardiac death. Therefore, to minimize cardiac risks the Food and Drug Administration (FDA) has mandated that in vitro cardiotoxicity screening be conducted early in drug development.69,70 Nonetheless, drug-induced torsades de pointes (TdP) has resulted in numerous preventable patient deaths and the costly withdrawal of marketed drugs. 66
Cardiomyocytes derived from HiPSCs appear to have similar properties to those of human heart cells in vivo, that is, expression of similar ion channels, action potentials, and pharmacologically appropriate responses to cardioactive drugs. They also exhibit cardiac myocyte morphology, including sarcomeric structures, and express a fetal gene expression pattern. 72 Today, drugs are routinely screened for cardiac safety by assessing their effects on cardiac ion channels (such as hERG), overexpressed in tumor-derived cell lines. 66 This approach is limited, as the cell lines do not replicate completely the full complexity of the cardiomyocyte physiology. Evaluating drugs for cardiac safety in vivo in rodents is also limited due to species differences.64,73,74
Multiple approaches in vitro have been reported to evaluate a compound’s action on HiPSC-derived cardiac myocyte function, including electrophysiology, intracellular Ca2+ ion levels, and myocyte contractility. 63 Planar patch-clamp arrays, including multielectrode arrays (MEAs), have been used for recordings from HiPSC-derived myocytes.63,66,72 The duration of the field potential correlates with patient QT prolongation. 68 Optical imaging technologies, together with Ca2+ ion biosensors, have been employed to measure Ca2+ ion transients in HiPSC-derived myocytes in response to drug actions. 65 Optical imaging has been used to measure beat rate of the HiPSC-derived myocytes. 72
Ribeiro et al. 72 monitored single HiPSC-derived cardiomyocytes in arrays in which the individual myocytes were placed in wells containing fluorescent microbeads. Using video imaging, they observed bead displacement as a measure of single myocyte beating and so calculated mechanical output of the imaged cells. They also detected changes in sarcomeric length, in the individual myocytes, in relationship to myocyte beating. As the single cells could be micropatterned on HTS platforms, this provided a unique approach to screen large numbers of compounds for cardiac effects.
Recently, Sakai et al. 75 developed a co-culture of HiPSC-derived cardiomyocytes with rat sympathetic neurons. Here, they monitored the functional connectivity using MEAs, showing that the frequency of myocyte beating increased upon sympathetic nerve stimulation. In the future, one might expect that the rat sympathetic neurons will be replaced with HiPSC-derived neurons in co-culture, thereby providing a unique in vitro human heart system with a neuronal innervation to monitor drug cardiotoxicity.
Pharmaceutical organizations have reported the use of HiPSC-derived cardiac myocytes to predict human cardiotoxicity. AstraZeneca and Johnson & Johnson researchers have reported screening studies with compound libraries on the electrical activity of HiPSC-derived cardiomyoctes. They showed a high degree of correlation in terms of cardiotoxicity, in comparison to known human safety data.76,77 Furthermore, researchers at GlaxoSmithKline compared the pharmacological responses of HiPSC-derived cardiomyocytes to drugs with the response of cardiomyocytes from dogs or rabbits to the same drugs, again showing that human cells were more predictive of in vivo patient cardiotoxicity. 78 Pharmacological analysis also showed that HiPSC-derived cardiac myocytes provide a good prediction of QT prolongation and arrhythmia over in vitro hERG testing. 78 These studies, and others, have led to the Comprehensive In Vitro Proarrhythmia Assay Initiative, which proposes to integrate the use of HiPSC-derived cardiomyocytes into existing guidelines when assessing novel drugs for cardiac safety. 80
HiPSC-Derived Myocytes for Toxicity Screening in Human Disease Models
HiPSC-derived models of cardiac disease have proved important for studying mechanisms of heart disease, including dilated cardiomyopathy (DCM), long QT syndrome (LQT), catecholaminergic polymorphic ventricular tachycardia (CPVT), LEOPARD syndromes, familial hypertrophic cardiomyopathy (HCM), familial DCM, TdP, and other reentrant arrhythmias.63–68,79,104,116 HiPSC-derived cardiac disease models can also be useful in evaluating drug toxicity, given that adverse cardiac drug reactions occur to a large extent in patients with hereditary cardiac disease. For example, episodes of TdP and sudden cardiac death related to cisapride were largely absent from the general population, but limited to patients with preexisting heart conditions, including LQT and heart failure.81,82 Patients with LQT, HCM, and DCM are particularly sensitive to cardiotropic drugs, and therefore vulnerable to fatal arrhythmias. 66 Given that there are relatively few methods to screen drug effects on human models of cardiac disease, the use of patient HiPSC-derived cardiomyocytes might result in improved safety.
Many of the human cardiac models show phenotypes and drug responses consistent with results from primary human tissues and patient testing. HiPSC-derived cardiomyocytes from a patient carrying the mutation that causes LQT exhibited a QT interval 170% longer than that of the control. 67 To characterize the iPSC-derived cardiomyocytes with LQT mutations, the cell culture model was tested against several ion channel blockers to establish the correlation of responses to the electrophysiological signature of LQT. 83 Treating LQT hiPSC-derived cardiomyocytes with potassium channel blockers exacerbated the LQT phenotype, whereas calcium channel blockers had an ameliorating effect, resembling the clinical effects of these drugs on LQT. 66 HiPSC-derived cardiomyocytes from patients with CPVT were characterized by their calcium handling in response to catecholamines. 63 Dantrolene, which rescued the CPVT phenotype in the knock-in mouse model, was also found to alleviate the abnormal delayed after-depolarizations seen in CPVT hiPSC-derived cardiomyocytes, reinforcing its possible therapeutic use for CPVT.
To further test if HiPSC-derived cardiomyocytes could model increased susceptibility of genetic cardiac disorders to drug-induced cardiotoxicity, Liang et al. 66 screened a panel of HiPSC-derived cardiomyocytes from patients with LQT, HCM, and DCM against compounds known to modulate cardiac ion channels. Patient-specific HiPSC cardiomyocytes from LQT, HCM, and DCM family cohorts expressed several cardiac disease phenotypes, including cellular hypertrophy and disorganization of sarcomere content. The HiPSC-derived cardiac myocytes from LQT patients had longer action potential durations. Finally, HiPSC-derived cardiac myocytes from LQT and HCM patients had a high incidence of arrhythmia, including early after-depolarizations and delayed after-depolarizations.
HiPSC cardiac myocytes derived from patients with cardiac diseases express different sensitivity to cardiotoxic drugs than cardiac myocytes derived from disease-free individuals, indicating that patient subpopulations exhibit different sensitivities to adverse cardiac drug reactions. Consequently, patient-specific HiPSC cardiomyocytes can be used to predict drug candidates with human cardiotoxicity better than methods presently available, providing an approach to exclude toxic compounds for further development before testing in humans.
Human Kidney Cells
Like the liver and heart, the kidney is also a key organ for drug toxicity, specifically the renal proximal tubule, due to its critical role in glomerular filtration and drug transport. 84 Numerous FDA-approved drugs cause nephrotoxicity. Moreover, identification of renal toxicity is frequently seen only in late clinical development, partly due to the limited predictability of preclinical animal nephrotoxicity models. Kandasamy et al. 85 showed that HiPSCs can be differentiated to form human renal proximal tubules. These cells were utilized in an assay to monitor renal toxicity based on increases in interleukin 6 and interleukin 8 expression. The cells were screened against 30 compounds known to induce varying levels of nephrotoxicity. The data from the studies showed that the HiPSC-derived cells provided an accurate readout of nephrotoxicity. These workers also showed that drug toxicity effects on HiPSC-derived human renal proximal tubules were consistent with known mechanisms of patient toxicity.
Consequently, HiPSC-derived human renal proximal tubules provide one of the first in vitro systems to assess potential human nephrotoxicity prior to human clinical testing. This provides a major benefit in profiling drugs for clinical development and may reduce the attrition rate in drug development due to kidney damage.
Human Neurons
HiPSCs can also be directed to differentiate into specific neuronal populations, including motor, cortical, dopamine, and striatal neurons.11–19,22–39 These cell phenotypes are of high interest in drug discovery, as they are most vulnerable in neurodegenerative diseases such as AD and FTD (cortical neurons),22–24,29–32 ALS (motor neurons),11,25,27,28 HD (striatal neurons),33,34 and PD (dopamine neurons).35–39 These neuronal types can be generated from patients with these diseases to mirror the pathophysiology, as well as to provide cells for HTS to identify neuroprotective agents. As with the other cell types discussed above, HiPSC-derived neurons can also be employed to screen for drugs for neurotoxicity potential.
Several methods have now been developed to generate pure populations of neurons from HiPSCs for use in HTS. 86 Several assay systems are also available to utilize HiPSC-derived neurons for drug discovery and to evaluate compounds for neurotoxicity, including MEAs to monitor electrical activity87–89 and imaging to monitor phenotypic responses, for example, neurite outgrowth and neuronal viability.11,14 HiPSC-derived cortical neurons 90 are compatible with HTS for electrical activity monitored using an automated patch-clamp instrument (IonFlux microfluidic system, Molecular Devices, Sunnyvale, CA, USA). The authors of this study employed a 96-well format and showed that the neurons expressed a robust and potent inhibitory response to GABAergic agonists with pharmacological selectivity consistent with responses seen in rodent neuronal cultures. These findings suggest that HTS programs using electrophysiological endpoints and HiPSC-derived neurons could be more commonly used in CNS drug discovery programs. These assays can also be used to identify novel CNS drugs that show selectivity to specific neuronal populations, particularly if one tests drug candidates against different neuronal populations (glutamate; GABA; dopamine plus cortical, striatal, and motor neurons) in the same assay format.
HiPSC-Derived Neurons for Toxicity Evaluation
As mentioned above, imaging technologies can be used to monitor the morphology of HiPSC-derived neurons in HTS formats for both screening and monitoring neurotoxicity. 91 These imaging technologies primarily focus on monitoring drug effects on the neurite area, probably because neurite outgrowth is critical for neuronal development, synapse formation, and cell–cell communication. Therefore, measuring changes in the neurite area provides an important readout on potential compound neurotoxicity. 92
To validate the utility of the neurite imaging assay using HiPSC-derived neurons for phenotypic toxicity screening, the assay was adapted into an HTS format and tested against a library of more than 4400 compounds. 92 It was observed that less than 3% of the compounds impaired neurite outgrowth of the HiPSC-derived neurons, with 37 being FDA-approved drugs known to target proteins or pathways involved in neurite extension. This “unbiased” phenotypic assay validated the use of HiPSC-derived neurons for the evaluation of compound liabilities with potential CNS effects. The data also suggest that hiPSC-derived neurons provide useful models to study the mechanisms of action (and off-target activities) of approved drugs, leading to a better understanding of their clinical efficacy and toxicity.
HiPSC-Derived Neurons for Drug Discovery
HiPSC-derived neurons can also be used to discover therapeutically useful CNS active compounds with novel mechanisms of action.11–14 Niedringhaus et al. 93 cultured HiPSC-derived neurons into arrays on releasable polystyrene microrafts to generate 1600 uniform, mobile neuron mini-cultures. Compared with conventional methods, microraft cultures exhibited improved neuronal viability and sample-to-sample consistency.
These authors validated the utility of these mini-cultures to screen for drugs affecting presynaptic function by using a lipid FM 5-95 (FM, lipophilic styryl dye) biosensor. They compared the synaptic activity of HiPSC-derived neurons from a patient with fragile X syndrome with controls using spinning disk confocal imaging. They showed that the fragile X neurons expressed impaired synaptic activity, similar to that found in cultures of neurons from mouse models of fragile X and consistent with previous findings that fragile X neurons have reduced presynaptic vesicles and synaptic transmission. 94 They then used the assay to screen for drugs to improve synaptic activity to treat fragile X and other diseases of the CNS involving impaired presynaptic function.
Multiple HTS formats have now been developed to screen small-molecule libraries against HiPSC-derived neuronal cultures to identity novel disease-modifying drugs to mitigate neurodegenerative diseases.11–13 This is important given the growing population of patients that suffer from AD, PD, and other CNS diseases and for whom no treatments currently exist to abrogate disease progression. The availability of HiPSCs from patients with these conditions now provides a means to identify compounds that may selectively retard neuronal degeneration. Importantly, while these diseases differ considerably in their symptomology—and the neuronal populations most vulnerable to degeneration—there may also be commonalities in the neurodegenerative mechanisms. Indeed, it is possible that drugs that reduce neurodegeneration in one disease may be useful in treating others.
As neurodegeneration is a slow process, technologies have been developed to longitudinally visualize neurons over time to monitor disease progression. Rather than the static detection assays employed in most HTS formats in which neurons are studied at a single time point, automated robotic microscopy (RM) methods have been developed to provide a time-lapse picture of human neurons over a prolonged period.11,13,14,25,26,33,35 It has been used to study basic mechanisms involved in neurodegeneration and to quantify how drugs may slow the disease process in an HTS format.
RM instrumentation consists of an automated platform that daily moves cell plates from an incubator to the microscope, positioning the wells identically each time.11,14 When fluorescent markers, such as green fluorescent protein, are expressed in neurons, RM can be used monitor the same neuronal population daily over several weeks. RM resolution is such that within each well, the microscope can visualize thousands of individual neurons, providing high statistical power in analyzing population changes. The ability to analyze individual neurons in a population is important because neurons tend to degenerate at different rates; RM can distinguish differences in rates of death of adjacent neurons in the same well. Using promoters to selectively drive expression of the fluorescent probes in neurons, RM can monitor the fate of neurons in mixed cell populations, allowing for neuronal culture analysis of cells expressing astrocytes needed for the maintenance of function.
HiPSC-Derived Neuronal Co-Cultures
Neurons and Astrocytes
Although pure populations of human neurons are preferred for drug discovery, neurons depend on association with a range of other cell types for function and survival; astrocytes and glia, for example, provide critical metabolic support for neurons. Kayama et al. 87 and Kuijlaars et al. 89 have both described methods to co-culture HiPSC-derived neurons with astrocytes. Using Ca2+ imaging, electrophysiology, and confocal imaging, Kuijlaars et al. 89 showed that co-culturing HiPSC-derived neurons and primary human astrocytes supported maturation of neuronal network synchronization, synaptic connectivity, and enhanced neuronal survival when compared with pure populations of neuronal monocultures. This is consistent with the role of astrocytes in supporting neuronal function and longevity. The co-cultured cells expressed GABAergic and glutamate markers and responded to inhibitors of both neurotransmitter pathways. Kayama et al. 87 showed that co-culturing with astrocytes enhanced the frequencies of burst spikes and the synchronization of spiking patterns in the neurons as recorded by MEA. These findings are consistent with normalization of neuronal circuitry within the neuronal cultures. In summary, employing co-cultures of HiPSC-derived neurons and astrocytes provides optimal physiological systems to monitor the functional properties of human neurons and their potential use in compound screening.
Neuromuscular Junction
A key role of neurons is the control of end-organ function. Human movement depends on neurons projecting from the spinal cord to innervate skeletal muscle, and ultimately the firing activity of the motor neurons coordinates skeletal muscle fiber contraction. During aging or in diseases such as ALS, these control mechanisms decline. Co-culture organoid approaches to model the human neuromuscular junction can therefore be used to understand fundamental aging mechanisms or diseases that impair motor movement, as well as to discover drugs to improve motor control.
Demestre et al. 18 developed culture conditions such that HiPSC could be differentiated into myoblast-like cells that fuse and form multinucleated striated myotubes that phenotypically express key skeletal muscle markers. The muscle fibers contracted in response to both electrical stimulation and acetylcholine. They then showed that co-culturing the muscle fibers with motor neurons differentiated from HiPSCs resulted in the clustering of acetylcholine receptors and formation of neuromuscular junctions in the fibers.
Steinbeck et al. 16 developed functionally active motor neuron/skeletal muscle co-cultures, using human embryonic stem cell-derived motor neurons and primary human myoblasts. To establish functional connectivity between neurons and muscle cells, they employed optogenics. Here, using lentiviral vectors, they expressed in the motor neurons channel rhodopsin2-EYFP under control of a human synapsin promoter. This allowed activation of the motor neurons with blue light and subsequent monitoring of muscle cell contraction by video imaging and stimulation of Ca2+ ion transients. Importantly, they showed that excitation–contraction coupling was blocked by antibodies from a myasthenia gravis patient and other autoimmune disease lesions that disrupt neuromuscular junction activity.
These studies may therefore provide a platform to study the neuronal control of muscle contraction that could be adaptable into HTS formats for the discovery of novel drugs for treating ALS, a neurodegenerative disease involving the loss of neuromuscular junctions and CNS control of motor function.16–19,25,26 Furthermore, since motor control gradually deteriorates during aging, the assay could help identify novel drugs that slow the loss of elderly motor control, as well as reducing the muscle wasting that results from loss of neuronal innervation. Most importantly, since there are considerable species differences in motor neuron control of skeletal muscle, this approach may provide the first neuronal end-organ system to study and control human large muscle contraction.
To further define culturing systems to facilitate neuron–muscle contact, Takayama and Kida 95 photolithographically constructed two cell culture chambers, connected by 20-micron tunnels, in which HiPSC-derived central or peripheral neurons were grown. Axons were then allowed to grow from one chamber to the other and thus form neuronal contacts that were monitored immunohistochemically. Human neurons were functionally active (as assessed by Ca2+ ion imaging), and electrical stimulation of one set of neurons induced activation of the neurons in the second chamber, indicating the formation of functional cell–cell contacts. Using this approach, the authors placed HiPSC-derived peripheral neurons in one chamber and cardiac myocytes in the other, and morphological and functional neuron–muscle contacts were established.
Consequently, it is now possible to engineer neuronal circuitry and neuromuscular synapses for functional monitoring. This system could facilitate studies to understand how neuronal circuitry is disrupted in diseases such as AD, and how neuron–organ connectivity can be compromised in diseases such as ALS. Indeed, the technology could be adapted for HTS to identify compounds specifically designed to affect the activity of targeted neuronal circuits underlying neurodegeneration.
HiPSC-Derived Models of Human Neurodegenerative Diseases
Since HiPSCs can be generated from patients with many different diseases, they may provide culture models ( Table 1 ) to investigate fundamental mechanisms involved in human pathology, and provide a source of cells for screening programs to identify novel therapeutics.
Motor Neuron Disease
HiPSCs have been used to model several motor neuron diseases, including ALS and spinal muscular atrophy (SMA).11,19,25,26 ALS is a progressive neurological disorder characterized by weakness, muscle atrophy, and respiratory failure, due to motor neuron degeneration. No treatment is available that reduces the neurodegeneration or blocks disease progression. Critical mutations in several genes cause familial ALS, with some of them (as well as their protein products) contributing to the sporadic forms of the disease, which is the most prevalent form of ALS. 96 HiPSC-derived motor neurons have been generated from ALS patients with familial disease, including those expressing mutations in the TDP43, C9ORF72, SOD1, and FUS genes. 96
TDP43 is an RNA regulatory protein and mutations in the TDP43 gene cause ALS, resulting in impaired transport of the protein to the nucleus, which in turn diminishes TDP43’s ability to regulate RNA processing. 97 Pathological cytoplasmic aggregation of TDP43 is found in patients with familial ALS, as well as in patients with sporadic ALS, suggesting that dysfunction of the protein is a major causative factor in the disease. Furthermore, HiPSC-derived motor neurons from ALS patients with TDP43 mutations recapitulate the cytoplasmic TDP43 aggregation found in patients. In support of this, Egawa et al. 98 generated iPSCs from three ALS patients carrying the TDP43 mutation and found that iPSC-derived motor neurons displayed reduced axon length and greater sensitivity to cellular stressors. Bilican et al. 25 reported that motor neurons differentiated from HiPSCs from ALS patients with the M337V mutation also exhibited TDP43 pathology, neurodegeneration, and reduced survival times. By adapting the HiPSC-derived motor neurons into an HTS format, these authors showed that compounds that increased autophagy to increase clearance TDP43 rescued the disease phenotype of ALS motor neurons.
HiPSC-derived motor neurons have also been developed from ALS patients expressing the C9ORF72 mutation, which is a GGGGCC repeat expansion in a noncoding region of the chromosome 9 open reading frame 72 gene. 11 The C9ORF72 mutation is the most common mutation known to occur in ALS as well as in FTD.11,96 While the mechanisms by which the C9ORF72 mutation causes familial ALS are unknown, individuals with the mutation present substantial TDP43 pathology, particularly TDP43 aggregates in the CNS. Consequently, the mutation may facilitate the pathogenic nature of other proteins linked to ALS, such as TDP43, and may suggest a broader role of TDP43 in ALS beyond those patients with TDP43 mutations.
In fact, Burkhardt et al. 99 showed that HiPSC-derived motor neurons from ALS patients with sporadic disease express TDP43 aggregation, and the aggregation recapitulates pathology in postmortem tissue from the patients from which the iPSCs were derived. These authors developed an HTS assay using diminished TDP43 aggregation in motor neurons as an assay endpoint. They identified FDA-approved compounds that reduced aggregation, supporting the use of this assay to identify TDP43-targeted drugs. These findings are of interest because TDP43 aggregation and pathology has also been identified in AD and FTD, two of the most common neurodegenerative diseases. Thus, TDP43-directed therapeutics may be useful in slowing neurodegeneration in both familial and sporadic ALS patients, as well as those afflicted with dementias.
Altered activity of motor neurons has been proposed to contribute to neurodegeneration in ALS. Nerve conduction studies evaluating the axonal threshold in ALS patients demonstrate increased axonal membrane excitability, and the degree of hyperexcitability correlates with patient survival. 100 Increased persistent sodium or reduced delayed-rectifier potassium currents may be responsible for the axonal hyperexcitability 100 and further contribute to neurodegeneration in this disease. The enhanced neuronal activity may contribute to intracellular calcium overload to impair mitochondria function, much like what has been proposed in the neurodegeneration of dopamine neurons in PD.
Studies by Wainger et al. 101 showed that HiPSC-derived motor neurons from ALS patients with the C9ORF72 mutation, as well as mutations in SOD1 and FUS, which also cause familial ALS, exhibit hyperexcitability. ALS-derived motor neurons show reduced delayed-rectifier voltage-gated potassium currents compared with controls. They found that retigabine, an activator of Kv7 potassium channels and a drug approved to treat epilepsy, reduced excitability to levels seen in controls and increased the survival of SOD1A4V/+ ALS motor neurons. These findings suggest that hyperexcitability may contribute to motor neuron degeneration in ALS. The blockade of hyperexcitability by retigabine was shown across several motor neuron types derived from familial ALS patients harboring different mutations, suggesting that the altered activity is common in this disease. Therefore, drugs targeting this hyperexcitability may slow neurodegeneration to treat ALS. In fact, retigabine is currently being tested in placebo control phase II clinical trials with ALS patients to establish the safety and efficacy in reducing disease symptoms (https://www.clinicaltrials.gov/ct2/show/NCT02450552).
In addition to ALS, HiPSCs have been employed to model SMA. 102 SMA is a motor neuron disease characterized by the degeneration of the anterior horn cells of the spinal cord, which subsequently leads to skeletal muscle atrophy and weakness. In SMA, pathological changes of the neuromuscular junction precede the motor neuron loss. To model the disease, Yoshida et al. 102 generated HiPSC-derived motor neurons from SMA patients and then co-cultured the neurons with the murine skeletal muscle cell line C2C12. They monitored the expression of neuromuscular junction using alpha-bungarotoxin staining, a marker of acetylcholine receptor clustering at neuromuscular junctions. Alpha-bungarotoxin staining was greatly reduced in the co-cultures of HiPSC-derived motor neurons from the SMA patients compared with controls. These authors 102 then showed that the drug valproic acid increased acetylcholine receptor clustering, indicating that the disease phenotype could be pharmacologically rescued. Valproic acid is known to increase the expression of SMN protein, which is downregulated in SMA due to mutation in the SMN1 gene. This co-culture system provides a simple format for HTS of compounds that may be useful in treating SMA and could likewise be used for drug discovery for other motor neuron diseases, like ALS.
In addition to studying the neuronal component of neuromuscular junctions, HiPSC can be directed to differentiate to skeletal muscle, which can be used for drug screening. Swartz et al. 103 developed methods to generate multinucleated skeletal myotubes using eight independent iPSC lines from FTD patients with the C9ORF72 mutation and sporadic FTD patients and controls. The skeletal muscle cells exhibited spontaneous contractions and could be adapted to an HTS format using imaging technologies to monitor contractions as well as survival. Thus, HiPSC-directed myocytes could be used to discover drugs targeting skeletal muscle to treat motor neuron diseases as well as skeletal muscle diseases such as Duchenne and Becker muscular dystrophies.
Alzheimer’s Disease
AD is the most common neurodegenerative disease, affecting about 30 million people globally. Like other neurodegenerative diseases, familial forms of AD are caused by several different gene mutations that contribute to neurodegeneration. While extensive studies over the last 20 years have focused on developing therapeutics to treat AD, no therapy has yet been identified to slow disease progression. This is due, in part, to the use of suboptimal animal disease models that fail to recapitulate the disease, but also to the lack of models of sporadic AD—the most prevalent form of the disease. HiPSC cultures have been used to study disease mechanisms in AD and provide models of the human disease for drug discovery.
Wang et al. 15 generated pure populations of cortical glutamate neurons from HiPSCs. These neurons expressed action potentials and spontaneous postsynaptic currents that were blocked by glutamate receptor antagonists. The neurons were used in HTS assay formats to screen for compounds that selectively lowered the levels of tau, a protein linked to AD neurodegeneration. In proof-of-principle studies, they identified a class of adrenergic drugs that selectively and effectively lowered tau production in HiPSC-derived neurons.
Studies by Kondo et al. 24 generated neurons from patients with familial AD and patients with trisomy 21/Down syndrome, a condition that overexpresses A beta, a peptide involved in AD neurodegeneration. Here, neurons were incorporated into an HTS format and screened against compound libraries. Molecules were identified that lowered A beta expression. Kondo et al. 24 used the assay format to prioritize “hits” by structure-based clustering to select lead compounds and further showed that the leads selectively reduced A beta expression in AD patient-derived neurons.
Ochalek et al. 105 generated neurons from HiPSCs of AD patients with sporadic disease, as well as those with familial disease caused by PSEN1 gene mutation. They showed that the neurons from the sporadic and familial AD patients both had increased levels of tau and phosphorylated tau. Phosphorylation is a posttranslational event that increases the tau toxicity in AD. Furthermore, a kinase linked to tau phosphorylation, GSK3B, was increased in neurons from patients with both forms of the disease. These data suggest that human AD iPSC-derived neurons could be employed in screening programs to identify small molecules that reduce tau phosphorylation to mitigate the disease, and so slow neurodegeneration. HiPSC-derived neurons from patients with sporadic AD thus provide a unique source of cells to discover novel agents targeting most AD patients.
Like ALS, neuronal hyperexcitability may also be involved in the pathogenesis of AD and may precede the degeneration seen in this disease.106–110 In AD, functional imaging of patients with mild cognitive impairment and early AD shows increased hippocampal hyperactivity. Transcranial magnetic stimulation of patients with early signs of AD shows hyperexcitability of cortical neurons, and epileptiform seizure activity is often found in AD patients before overt neuronal loss or cognitive decline. Several studies have shown that tau is a critical protein involved in AD pathogenesis; iPSC-derived neurons from patients with FTLD-tau exhibit dysregulated electrically evoked Ca2+ ion transients. Blocking this hyperactivity reduced tau accumulation and neurodegeneration. 31 Murine models of AD also show epileptic activity prior to overt pathology, and the antiepileptic drug levetiracetam reduced abnormal spike activity and reduced hippocampal remodeling, behavioral abnormalities, synaptic dysfunction, and deficits in learning and memory. 109 These observations further support the hypothesis that aberrant network activity contributes causally to synaptic and cognitive deficits in AD, and that levetiracetam could reduce the risk of individuals for AD. In this regard, levetiracetam has been reported to diminish network dysfunction in patients with amnestic mild cognitive impairment. 111
Importantly, both HiPSC-derived neuronal models of ALS and AD exhibit hyperactivity prior to neuronal loss.27,28,31 Since methods have been developed to use MEAs to monitor neuronal activity in HTS formats, screening programs could be developed to identify compounds that selectively reduce this hyperactivity in the disease models. This may provide the basis for developing new drugs to normalize the activity of the vulnerable neurons to reduce neurodegeneration and thus slow the progression of ALS, AD, and potentially other degenerative diseases, such as PD, a disease in which hyperactivity dopamine nigrostriatal neurons is linked to neurodegeneration and disease progression.
Parkinson’s Disease
PD is the second most common neurodegenerative disease.35–39 The disease is progressive, with motor and nonmotor disabilities increasing over time. While motor symptoms of PD can be treated with carbidopa and, in some cases, deep brain stimulation, the efficacy of these therapies diminishes over time. Currently, there is no therapy that slows the progression of PD.
Mutations in several genes are linked to PD, including those for alpha-synuclein (aSYN) and LRRK2, which cause familial forms of PD and contribute to sporadic forms of the disease.35–39 aSYN is a prime risk factor for disease progression in all PD patients. 112 One of the first HiPSC models of PD involved the generation of dopamine neurons from PD patients possessing the aSYN triplication mutation. 113 The neurons expressed increased levels of aSYN and increased sensitivity to oxidative stressors that induced neurodegeneration. HiPSC-derived neurons from patients with other disease-causing aSYN gene mutations have also been generated, including those with the A53T mutation.
LRRK2 mutations are the most common genetic cause of PD.35,38 The most frequent mutation, particularly in individuals of European descent, is the G2019S mutation. The LRRK2 (G2019S) mutation is found in almost 1% of all PD patients worldwide. A wider role for LRRK2 in PD comes from genome-wide association studies (GWAS), showing that polymorphisms near the LRRK2 gene increase risk of sporadic PD. Mutant forms of LRRK2, including the LRRK2 (G2019S) mutation, induce neurodegeneration in cell-based and animal models of PD. 35 HiPSC-derived neurons from PD patients with the G2019S mutation have been studied by multiple groups.35–38 These models show increased sensitivity to oxidative stress, mitochondria damage, reduced neurite area, and increased risk of death. Several studies have also directed the HiPSC to tyrosine hydroxylase-positive neurons with a dopamine phenotype, the cells most vulnerable in PD, and the patient neurons exhibit accelerated neurodegeneration compared with the control.35,38 Disease onset and symptoms of sporadic PD patients and those harboring the LRRK2 (G2019S) mutation are similar, suggesting that approaches to reduce LRRK2 (G2019S)-induced neurodegeneration will also help to slow the progression of disease in sporadic PD patients. Although carriers of the mutant gene LRRK2 (G2019S) are rare, LRRK2 has a broader role in disease PD pathogenesis since LRRK2 (G2019S) interacts with aSYN to cause neurodegeneration.
The interplay between LRRK2 and aSYN is supported, in part, by studies using HiPSC. 35 Here, the HiPSC-derived neurons, from PD patients with the G2019S mutation, exhibited increased expression of aSYN, and knockdown of aSYN is protective of the dopamine neurons. Increased expression of aSYN is due to impaired clearance because LRRK2 is an inhibitor of autophagic flux, a major pathway to clear misfolded proteins such as mutant LRRK2 and aSYN. The HiPSC-derived dopamine neurons from patients with aSYN and LRRK2 mutations, as well as patients with sporadic disease, thus provide a rich source of human models for adaptation in HTS formats to identify novel chemical leads to reduce the degeneration of dopamine neurons in PD to slow progression.
Huntington’s Disease
HD is an autosomal dominant neurodegenerative disease caused by an expansion of CAG repeats in the huntingtin (htt) gene.33,34 The length of the expansion varies and correlates with disease severity. With extreme CAG expansion, symptoms develop in childhood, the pathology is extensive, and life is short. HiPSC-derived neurons from HD patients with varying CAG repeats have been developed. The most extensively studied neuronal population is HiPSC-derived striatal cells since they are in the brain region most affected in the disease. 33 The first HiPSC striatal neurons maintained the genetic mutation and exhibited multiple disease-relevant phenotypes, including expression of mHtt aggregates, impaired electrical activity and action potential generation, and increased risk of death. 33 Using imaging technologies, HD striatal neurons were shown to have impaired turnover of mHtt, which resulted in increased expression of the disease-causing protein. 114 The altered turnover of mHtt was statistically predictive of neurodegeneration. The HD neurons were then incorporated into an HTS assay format that monitored neuronal survival over time. Through screening of the HD neurons with a library of FDA-approved drugs, several commonly used drugs that increase autophagy in the HD neurons and reduce neurodegeneration were identified. 115 These studies, and others, consequently support the development of autophagy inducers to treat HD using HD neurons.
Direct Expression of Human Neurons from Fibroblasts
In general, HiPSC-derived neurons express the phenotype of embryonic, immature neurons, probably because the reprogramming removes that epigenetic markers expressed in the patient. Consequently, the molecular context of the patient’s cells is lost. While this phenomenon provides a direct readout of the genetic mutation effect on the cell phenotype, it removes other characteristics of the patient’s cells that contribute to disease, such as the patient’s age. To overcome these problems, technologies have now been developed to directly generate human neurons from patient fibroblasts, without the need to produce iPSCs.40–43
Yoo and colleagues40–42 used miRNAs to directly convert human fibroblasts to neurons. The neurons maintained their age-associated signatures, including the extent of DNA damage and telomere length from which the fibroblasts were obtained. Striatal neurons could be differentiated directly from fibroblasts of HD patients and were shown to exhibit disease phenotypes, including aggregates of mHtt, mHtt-induced DNA damage, and mitochondrial damage and spontaneous degeneration over time. These studies, collectively, suggest that it is possible to generate neurons from patient fibroblasts that retain both the epigenetic marks and the patient’s genetic mutation.
Loss of a patient’s epigenetic background is one reason why HiPSC-derived neurons derived from patients with sporadic forms of other neurodegenerative diseases lack robust phenotypes. Using the approach developed by Yoo and colleagues40–42 therefore provides a better way to study the neurodegenerative process in patients with sporadic (and more prevalent) forms of CNS diseases, including ALS. Notably, Tang et al. 43 reported that motor neurons, generated directly from human fibroblasts, maintained many molecular characteristics of the aged donors, such as extensive DNA damage, loss of heterochromatin, and nuclear organization. Therefore, this, and other approaches, may provide a means to visualize the heterogeneity of the molecular and physiological phenotypes of patients with sporadic forms of neurodegenerative diseases, providing a platform to stratify those patients that may be effectively treated with novel therapeutics, prior to clinical testing.
Summary
The use of HiPSCs is now impacting drug discovery programs, facilitating the identification of novel compounds to treat a range of human diseases. The versatility and patient relevance of HiPSCs provide many advantages for drug discovery, including disease modeling, target and lead identification, and lead optimization.
When cells are cultured from the same individual, one can now reproducibly culture many different cell types in high abundance. This clearly aids in identifying compounds with desired biological effects, but it also provides for optimization of compounds in terms of efficacy, selectivity, and safety. Furthermore, HiPSCs are particularly important given that animal models of many human diseases either do not exist or are poorly reflective of the disease pathology. This is perhaps most apparent in neurodegenerative diseases for which no animal model exists for any sporadic forms of these diseases. This is one reason why no drugs have been developed that slow neurodegenerative disease progression.
Finally, HiPSCs will find widespread utility in preclinical safety testing, given that in vitro and in vivo safety evaluation has historically been conducted using animal-derived cells or in vivo models, many of which lack predictability in the clinic. This is most apparent in screening compounds for cardiovascular safety, but it is also increasingly the case for predicting metabolic liability in the liver. Lead optimization can now be undertaken in cells both from healthy individuals and from patients with different diseases, thereby selecting compounds with a lower risk of side effects in the specific patient population.
Taken together, the result may well be the discovery of a new generation of drugs more effective in treating diseases that in many respects are still untreated.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.