
Abstract
Dipeptidyl peptidase 1 (DPP1) (EC 3.4.14.1; also known as cathepsin C, cathepsin J, dipeptidyl aminopeptidase, and dipeptidyl aminotransferase) is a lysosomal cysteinyl protease of the papain family involved in the intracellular degradation of proteins. Isolated enzyme assays for DPP1 activity using a variety of synthetic substrates such as dipeptide or peptide linked to amino-methyl-coumarin (AMC) or other fluorophores are well established. There is, however, no report of a simple whole-cell-based assay for measuring lysosomal DPP1 activity other than the use of flow cytometry (fluorescence-activated cell sorting) or the use of invasive activity-based probes or the production of physiological products such as neutrophil elastase. The authors investigated a number of DPP1 fluorogenic substrates that have the potential to access the lysosome and enable the measurement of DPP1 enzyme activity in situ. They describe the development and evaluation of a simple noninvasive fluorescence assay for measuring DPP1 activity in fresh or cryopreserved human THP-1 cells using the substrate H-Gly-Phe-AFC (amino-fluoro-coumarin). This cell-based fluorescence assay can be performed in a 96-well plate format and is ideally suited for determining the cell potency of potential DPP1 enzyme inhibitors.
Introduction
D
COPD is characterized by a slowly progressive and irreversible deterioration in lung function leading to airflow limitation, as confirmed by spirometry. 4 A number of proinflammatory cells are associated with the pathogenesis of COPD, but elevated neutrophil numbers in the airways and sputum of COPD patients are a well-recognized pathological feature. 5,6
The association of DPP1 with the pathogenesis of COPD has provided an impetus to find novel inhibitors of this enzyme. Development of DPP1 inhibitors by Arpida, Prozymex, Bayer, Merck, GlaxoSmithKline (GSK), Novartis, and AstraZeneca has been reported, and it is apparent that some of these potent DPP1 inhibitors have poor drug metabolism pharmacokinetic (DMPK) properties. 7-14 Furthermore, recent studies have predicted that very high DPP1 fractional inhibition will be required for successful therapeutic intervention in humans, which may make DPP1 a more challenging target for the treatment of COPD than first realized. 15,16
We have initially identified novel DPP1 inhibitors through our in-house high-throughput screening (HTS) program using a purified human recombinant DPP1 enzyme fluorescence assay. To pursue these DPP1 inhibitors, we needed to demonstrate cellular DPP1 activity because of the location of DPP1 within the cell lysosome. Hence, only good inhibitors with the right physicochemical properties will have access to the lysosome and inhibit in situ DPP1 activity. Finding such compounds with potent DPP1 enzyme and cell activities is critical for further progression in vitro and subsequent testing in diseased animal models.
In this article, we describe the development and validation of a simple cell-based assay to measure lysosomal DPP1 activity. Previously, invasive or endpoint methods have been employed to assay lysosomal cathepsins using activity-based irreversible probes 17,18 or physiological protein products of DPP1 peptidase activity (e.g., elastase, cathepsin G, proteinase 3) 15,16 or cytotoxic products of DPP1 polymerase activity such as L-leucine methyl ester or L-leucine-leucine methyl ester. 19-21 In addition, the proteolysis of fluorogenic rhodamine peptidic substrates in B721 EBV-immortalized B cells using fluorescence-activated cell sorting (FACS) has been reported only very recently. 22 Unlike the cellular DPP1 methods as reported above, we have used the cell-permeable fluorogenic substrate, H-Gly-Phe-AFC, in conjunction with the human monocytic leukemia cell line, THP-1, to design a simple DPP1 cell assay for screening potential DPP1 inhibitors. We have chosen THP-1 cells because they contain high levels of DPP1, grow easily in standard laboratory conditions, and are good model cells for promyelocytes. Our in-house compound screening program has generated data with good correlation between the purified human recombinant DPP1 enzyme and cell assays and allowed us to further pursue these novel DPP1 inhibitors. Furthermore, this cell DPP1 assay is highly amenable to HTS.
Materials and Methods
Materials
All assay reagents and cell growth medium were obtained from Sigma-Aldrich UK (Dorset, UK), except for H-Gly-Phe-AFC, which was purchased from NeoMPS, Polypeptide Laboratories Group (San Diego, CA), and H-Gly-Arg-AMC, which was purchased from Bachem (Bubendorf, Switzerland). (Gly-Arg)2-rhodamine was synthesized in the Medicinal Chemistry Department at AstraZeneca R&D Charnwood (Loughborough, UK) from a GSK DPP1 inhibitor patent WO 2006094003. Human recombinant DPP1 (hrDPP1) was produced by the DECS Group at AstraZeneca R&D (Mölndal, Sweden).
Cultivation and cryopreservation of THP-1 cells
THP-1 cells, a human monocytic leukemia cell line, were originally obtained from American Type Culture Collection (TIB 202 F-11838; ATCC, Manassas, VA) and grown in RPMI + 1% (v/v) glutamine + 10% (v/v) fetal calf serum (FCS) at 37°C in a CO2 incubator. At near confluence, THP-1 cells in culture were harvested and resuspended into phosphate-buffered saline (PBS, pH 7.4) at the required density before being aliquoted into assay wells.
Cryopreserved THP-1 cells at a density of about 1.0 × 107/mL in 80% (v/v) of FCS, 10% (v/v) DMSO, and 10% (v/v) growth medium (RPMI) were stored at −150°C. Prior to the assay, cryopreserved cells were thawed in a water bath at 37°C, centrifuged at 300 g for 5 min to remove the cryopreservation medium, and then resuspended into PBS to the required cell density. Both fresh and cryopreserved cells used in the assay were usually >95% viable, as confirmed by the Cedex cell counter (Trypan blue uptake).
DPP1 enzyme assay
The activity of hrDPP1 was determined by measuring the enzymatic release of amino-methyl-coumarin (AMC) or amino-fluoro-coumarin (AFC) from their respective peptide substrates, H-Gly-Arg-AMC or H-Gly-Phe-AFC, leading to an increase in fluorescence intensity at Ex λ350 nm and Em λ450 nm for AMC or Ex λ400 nm and Em λ505 nm for AFC. The assay was carried out in black 384-well plates in a final volume of 50 µL at 22°C. The assay conditions contained the following: assay buffer (25 mM piperazine buffer, pH 5.0; 50 mM NaCl, 5 mM dithiothreitol [DTT]; 0.01% [v/v] Triton X-100), 100 µM H-Gly-Arg-AMC or H-Gly-Phe-AFC (~equivalent to Km), and hrDPP1 (~0.1 nM). Test compounds were solubilized in DMSO and then diluted in the assay buffer to give a final DMSO concentration of 1% (v/v). A 10-point half-log dilution series of the inhibitors was tested and the pIC50 determined using a 4-parameter logistic equation in a nonlinear curve-fitting routine. A standard irreversible and selective DPP1 inhibitor (vinyl sulfone [VS]) was used as an inhibitor control in the assay. 12 Routinely, compounds were preincubated with hrDPP1 for 30 min prior to the addition of the peptide substrate to start the reaction and then left for a further 60 min at 22°C. After this period, the plates were immediately read in a fluorescence plate reader using the above emission and excitation wavelengths. The Km for the substrate was determined using Michaelis-Menten-based plots.
DPP1 cell assay
The assay was carried out in a 96-well clear plate (Corning Costar; Corning, Lowell, MA). Then, 10 µL aliquots of compound (10-point half-log concentrations) or 10 µL of 5% (v/v) DMSO positive controls or 10 µL of VS (final assay concentration [FAC], 10 µM) negative controls were dispensed into the appropriate well, followed by 30 µL of 1.0 × 106/mL THP-1 cells in PBS added to all wells. The plate was preincubated at 37°C for 60 min in a cell culture incubator, and then 10 µL of H-Gly-Phe-AFC (FAC 100 µM ~equivalent to Km) was added to start the assay reaction. The plate was further incubated for 60 min at 37°C, and the product of the reaction was read in a fluorescence plate reader, Spectramax Gemini (Molecular Devices, Sunnyvale, CA; Ex λ400 nm, Em λ505 nm). There was little decrease in cell viability (<5%) within the experimental timescale. The potency (pIC50) of the test compounds in blocking the production of fluorescence product by THP-1 cells was determined using a 4-parameter logistic equation in a nonlinear curve-fitting routine.
The Km for H-Gly-Phe-AFC was determined using a Michaelis-Menten plot. A number of protease and lysosomal inhibitors, including bafilomycin A1, cystatin, elastase, cathepsin L, and cathepsin K, were profiled for selectivity in the DPP1 cell assay. For comparison, 2 other substrates, (Gly-Arg)2-rhodamine and H-Gly-Arg-AMC, were investigated in the THP-1 cells as described above. (Gly-Arg)2-rhodamine fluorogenesis was measured with Ex λ485 nm and Em λ535 nm.
Imaging of DPP1 in THP-1 cells using H-Gly-Phe-AFC
The assay was constructed in a 96-well Biocoat collagen type 1 plate (Becton Dickinson, Franklin Lakes, NJ). In a volume of 100 µL, the assay wells contained 10 µL of H-Gly-Phe-AFC (FAC 100 µM), 10 µL of DraQ-5 dye (FAC 2 µM; Biostatus, Leicester, UK), 10 µL of DMSO (FAC 1% [v/v]), and 70 µL THP-1 cells in PBS (i.e., final 56K cells). The plate was imaged on the ImageXpress 5000a (MDS Analytical Technologies, Sunnyvale, CA). Images were captured using a 20× PF ELWC objective. DraQ-5 was used because it is a nuclear marker spectrally distinct from the fluorescence of H-Gly-Phe-AFC. DraQ-5 was imaged using a λ700-75 nm emission filter with λ620-60 nm excitation and λ660 nm long-pass (LP) dichroic mirror. The exposure time for DraQ-5 was 250 ms. H-Gly-Phe-AFC fluorescence was captured using a λ535-50 nm emission filter, λ360-40 nm excitation, λ505 nm LP dichroic, and an exposure of 150 ms. Images of substrate H-Gly-Arg-AMC were similarly captured. Hoechst 33342 was used as a nuclear marker with the following acquisition settings: emission filter of λ460-50 nm, excitation of λ360-40 nm, λ400 nm LP dichroic, and an exposure of 50 ms. H-Gly-Arg-AMC images were acquired using an emission filter of λ620-60 nm, excitation of λ570-20 nm, λ585 nm LP dichroic, and 100-ms exposure.
Liquid handling
Throughout the experiments, liquid handling was carried out using ECHO 555 (Labcyte, Sunnyvale, CA) or Star Plus (Hamilton Robotics, Reno, NV) or handheld electronic pipettes (BIOHIT, Helsinki, Finland).
Results and Discussion
THP-1 cells were selected as a convenient cell assay system because they express high endogenous DPP1 activity 21,23,24 and are good model cells for promyelocytes, which are relevant to the biology of DPP1 expression. It was possible to use DPP1 transfected cells instead of THP-1 cells, but they would offer little advantage because of the difficulties in finding a mammalian host cell that has no endogenous DPP1 (hence may have 2 variant forms of DPP1 in the cell) and in generating a stable transfected cell line expressing active mature DPP1 in the lysosomes (i.e., DPP1 has a complex tetrameric structure and requires extensive processing and translocation of the zymogens to the active mature tetrameric enzyme). Another advantage of THP-1 cells is that they are a stable monocytic cell line that grow easily in standard laboratory conditions, hence providing a constant and consistent supply of cells for screening. Incubation of these THP-1 cells with 3 different fluorogenic substrates clearly demonstrated that H-Gly-Phe-AFC was the only substrate converted to its fluorescent product, whereas Gly-Arg-AMC and (Gly-Arg)2-rhodamine were not significantly turned over ( Fig. 1 ).
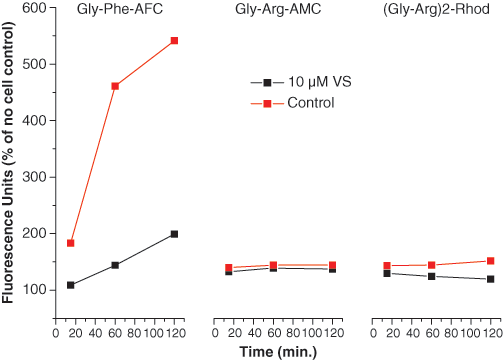
Cell fluorogenesis time courses for 3 different DPP1 substrates. THP-1 cells were incubated with 3 different DPP1 fluorogenic substrates in the presence and absence of 10 µM vinyl sulfone (VS), a selective DPP1 inhibitor, to determine the specific rate of product formation. Typical plots are presented where data points represent the arithmetic mean of 4 separate observations per condition.
We measured fluorogenesis initial rates at different cell ( Fig. 2A ) and substrate ( Fig. 2B ) concentrations. Cell numbers and H-Gly-Phe-AFC concentrations were adjusted such that the time course of fluorescent product generation was linear up to 1 h and for up to 200 µM substrate. The apparent Km for H-Gly-Phe-AFC in THP-1 cells was 111 µM ( Fig. 2C ), which is similar to the Km values (range, 75-270 µM) reported for the DPP1 enzyme assay using H-Gly-Phe-fluorophores. 10,14,25,26 Based on the above findings, the optimized conditions for inhibitor screening were chosen as 30K THP-1 cells, 100 µM H-Gly-Phe-AFC, and 60-min incubation time.
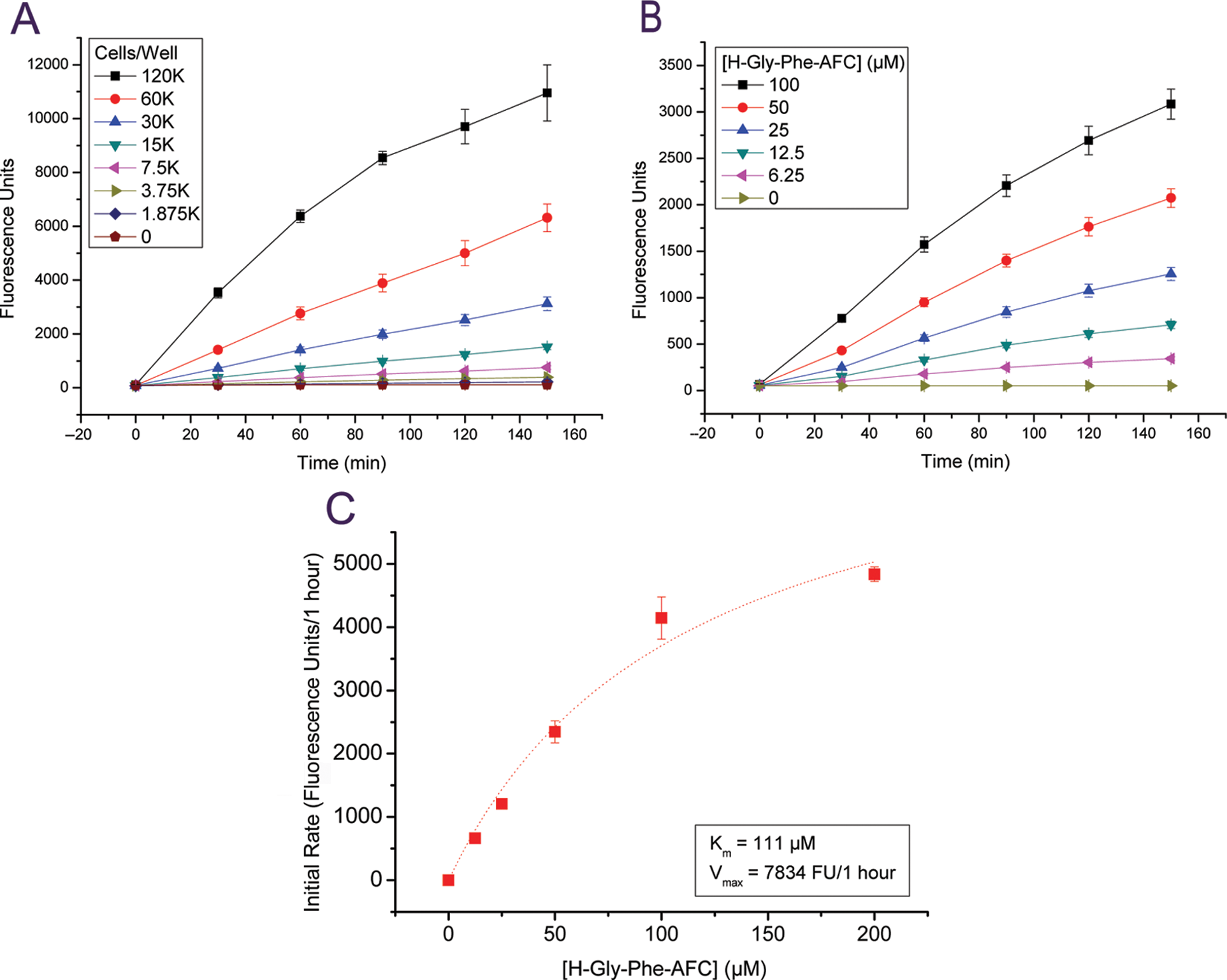
Kinetics of H-Gly-Phe-AFC fluorogenesis in THP-1 cells. (
However, a key question is whether DPP1 is solely responsible for the cellular turnover of H-Gly-Phe-AFC. A review of the literature suggests that the dipeptide H-Gly-Phe-fluorophore is a much better substrate for DPP1 12,14,25-27 compared with other proteases, and indeed H-Gly-Phe-CHN2 binds to the active sites of DPP1 as elucidated by the enzyme-crystal structure at 2.0 Å resolution. 28 We have demonstrated that bone marrow cell lysates from DPP1 knockout (KO) mice failed to hydrolyze H-Gly-Phe-AFC to any significant extent ( Fig. 3 ), suggesting that the cleavage of this substrate is mainly dependent on DPP1. Our results are in agreement with Pham and Ley, 29 who also showed that mouse KO bone marrow cell lysates lack the ability to hydrolyze H-Gly-Phe-β-naphthylamide. In addition, it has been demonstrated that natural killer (NK) cells from patients lacking DPP1 activity failed to hydrolyze the substrate H-Gly-Phe-para-nitroanilide. 30
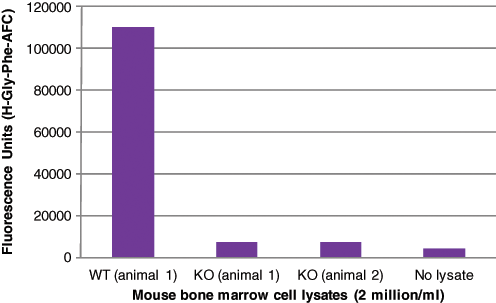
DPP1 activity from mouse bone marrow cell lysates. Bone marrow cells were extracted/isolated from wild-type (WT) and DPP1 knockout (KO) animals (backcrossed to C57BL/6(N6)) and lysed in lysis buffer (20 mM sodium acetate, 1 mM EDTA, and 1% [v/v] Triton X-100 pH 5.5 at 10 million cells/mL). DPP1 activity were measured from WT and DPP1 KO bone marrow cell lysates using the substrate H-Gly-Phe-AFC as described in the Materials and Methods section (DPP1 Enzyme Assay). The data represent the arithmetic mean of 2 separate observations per condition.
We further validated that cell turnover of H-Gly-Phe-AFC was due to DPP1 with a range of selective protease inhibitors and lysosomal reagents. VS is a potent selective irreversible inhibitor (pIC50 9.3) of DPP1 ( Table 1 and Kam et al. 12 ) that also inhibits cell turnover of H-Gly-Phe-AFC (pIC50 7.2), again supporting the essential role of DPP1 in this cell assay. In contrast, a range of other protease inhibitors for elastase, proteinase 3, cathepsin K, cathepsin L, DPPIV and urokinase, and a nonspecific serine protease inhibitor, phenylmethylsulfonyl fluoride (PMSF), were all inactive in the DPP1 cell assay, confirming that the cell turnover of H-Gly-Phe-AFC is most likely due to DPP1 activity.
Selectivity of Vinyl Sulfone (VS)
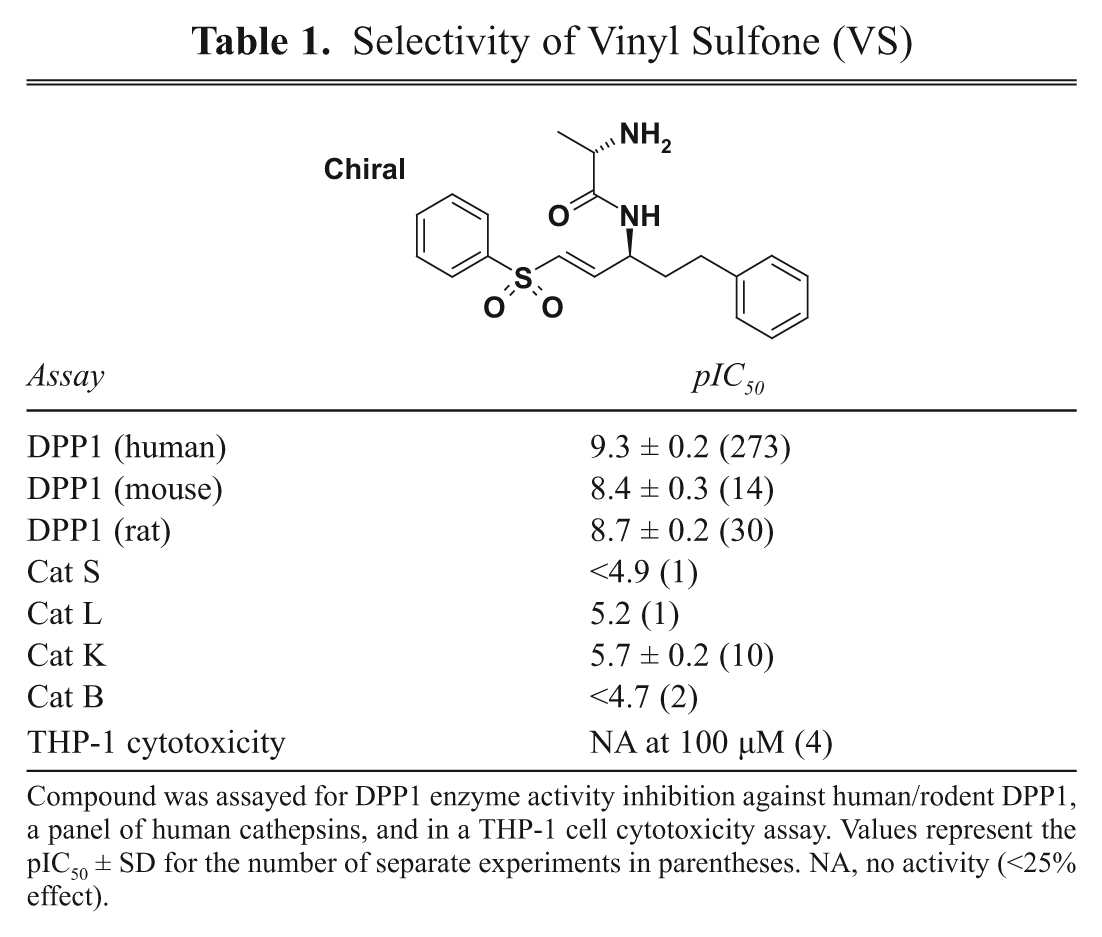
Compound was assayed for DPP1 enzyme activity inhibition against human/rodent DPP1, a panel of human cathepsins, and in a THP-1 cell cytotoxicity assay. Values represent the pIC50 ± SD for the number of separate experiments in parentheses. NA, no activity (<25% effect).
Cystatin, a 13-kDa cell-impermeable cysteinyl protease inhibitor (pIC50 8.3 for enzyme inhibition), was used to ascertain the cellular location of the DPP1 activity in the cell assay. Not surprisingly, cystatin was unable to block the turnover of the substrate in the cell assay ( Table 2 ), and this lack of inhibition suggested that the DPP1 activity resided within the cell and not externally, where it would be accessible to the inhibitory action of cystatin.
Pharmacology of the DPP1 Enzyme–Cell Assay Relationship

Compounds were assayed for human recombinant DPP1 enzyme and DPP1 cell assay inhibition, as described in Materials and Methods. Values represent the pIC50 ± SD for the number of separate experiments in parentheses. NA, no activity (<25% inhibition) at the concentration indicated.
The DPP1 cell assay, however, was potently inhibited by the vacuolar-type H+-ATPase inhibitor, bafilomycin A1 (pIC50 8.4), suggesting that blockade of lysosomal acidification attenuated DPP1 activity ( Table 2 ). It is likely that the acute cell cytotoxicity (pIC50 5.2 after 1-h incubation) of bafilomycin A1 may also contribute to the inhibitory activity seen in the DPP1 cell assay, as some known cytotoxins were shown to inhibit the cell assay without blocking DPP1 enzyme activity (data not shown). Therefore, pump inhibitors or cytotoxins must be discarded as cell assay false positives and can be identified by their relative inability to inhibit the isolated DPP1 enzyme activity. In addition, compounds that quench the cell assay fluorescence are also false positives and can be identified by their comparable ability to directly quench AFC (the cell assay product) fluorescence.
As part of a lead generation exercise to identify selective DPP1 inhibitors, emerging enzyme inhibitors were tested in the cell assay for attenuation of cellular DPP1 activity in situ ( Fig. 4 ). Here the cell assay measured the ability of the compounds to gain access to the lysosome and inhibit DPP1 activity in the presence of any endogenous substrates in its native environment. Within any given chemical series where members have similar gross physicochemical properties, the inhibition of enzyme activity correlated well with that for the cell assay, further confirming that the fluorogenesis in the cell assay was due to DPP1 activity. Generally, basic inhibitors tended to be better at blocking cellular DPP1 activity than acidic inhibitors, with some examples being inactive in the cell assay ( Fig. 4 ). A likely reason behind this is that basic drugs are better at permeating the cell membrane and accumulating in the lysosomes to inhibit DPP1 activity than acidic drugs. This profile is probably due to the well-known lysosomal accumulation of lipophilic bases (i.e., lysosomotropism) driven by the pH gradient that acidifies the lysosome.
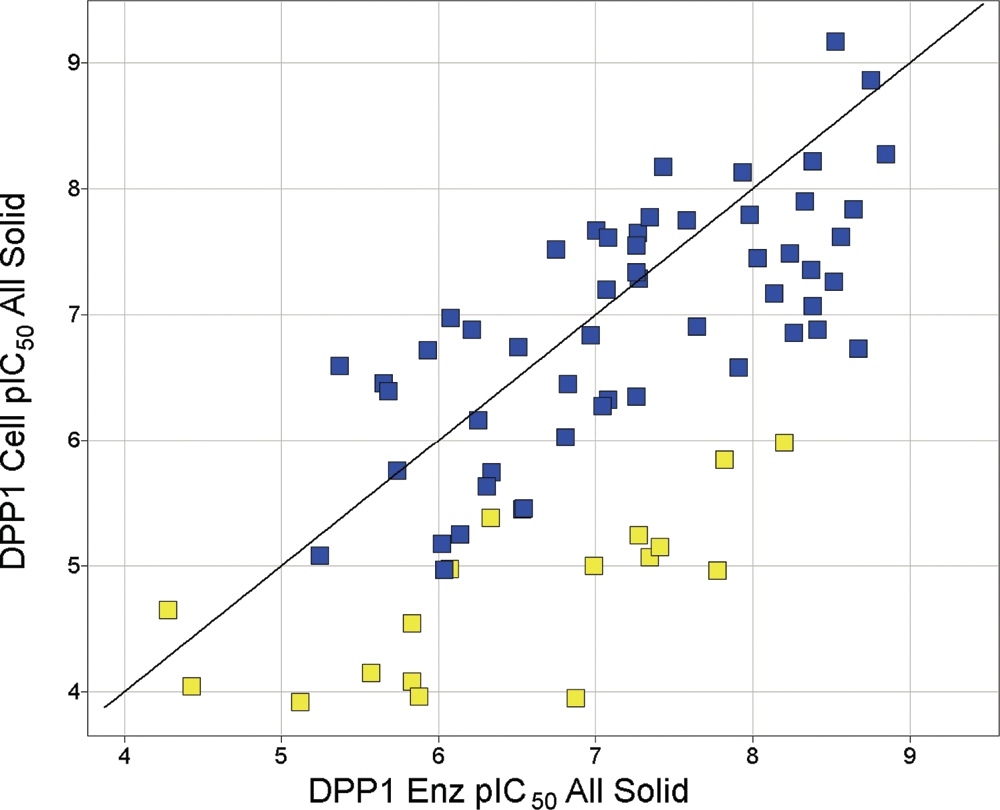
Plot of compound pIC50 for inhibition of DPP1 enzyme and cell activity. Data represent the mean pIC50 from 2 separate observations per condition for 71 compounds with similar lipophilicity (ACDLogD 0.5 to 1.5) tested in the DPP1 enzyme and cell assay. The line represents the line of unity, and acidic and basic compounds are labeled yellow and blue, respectively.
To investigate the intracellular site of the cellular DPP1 activity, we used subcellular imaging to locate fluorescent product formation (i.e., AFC) from H-Gly-Phe-AFC in THP-1 cells ( Fig. 5A ). At the cellular level, the formation of AFC (green) with time appeared to be concentrated in lysosomes with no nuclear staining (red), as shown by the overlay of the 2 different dyes ( Fig. 5B ). However, there was also some green staining in the cytoplasm that could be due to leakage of AFC out of the lysosome. On the other hand, the use of H-Gly-Arg-AMC did not produce any lysosomal staining (data not shown), suggesting that this substrate did not permeate into the cell, a finding consistent with the demonstration of the lack of fluorescent product formation in the cell assay ( Fig. 1 ). The inability of H-Gly-Arg-AMC to stain the cells may be due to the strong basic group (pKa ~12.5) in the arginine making the substrate highly zwitterionic, which led to poor passive cell permeability. However, there is a possibility that other factors beyond permeability could account for this finding and remains to be investigated.
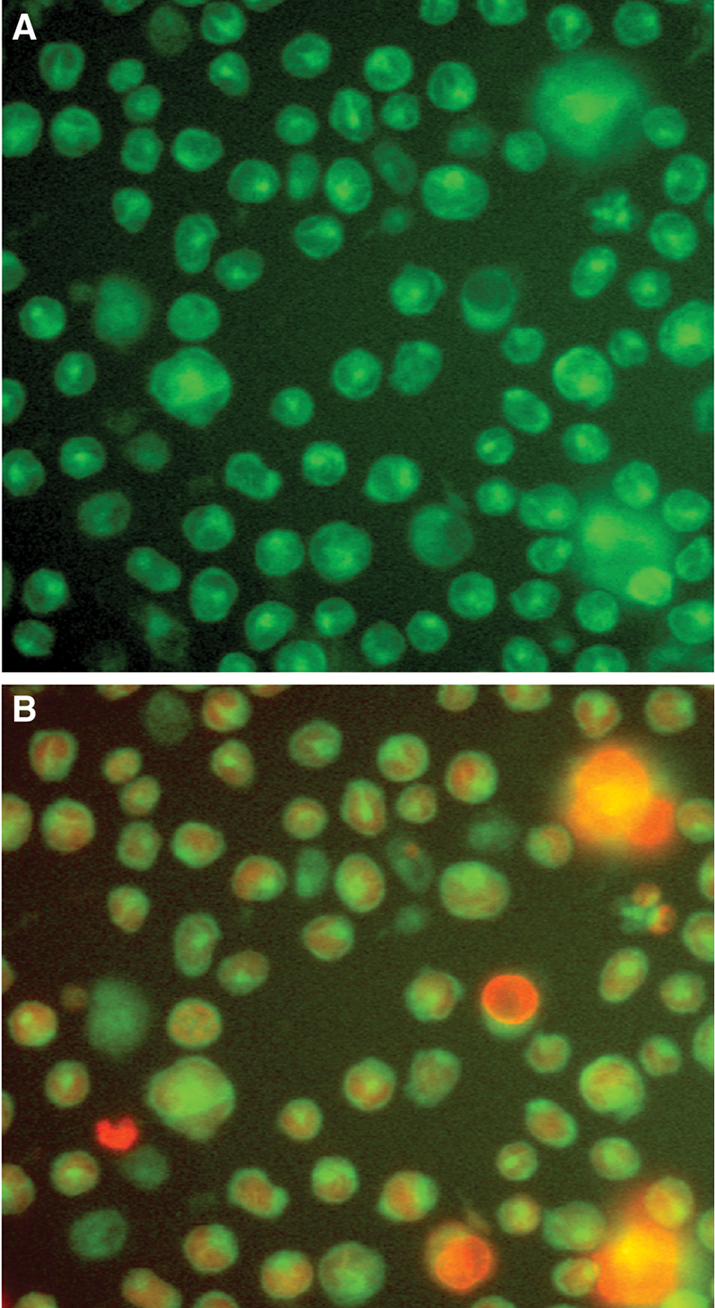
Imaging of THP-1 cells incubated with the DPP1 substrate H-Gly-Phe-AFC. THP-1 cells were incubated with the DPP1 substrate H-Gly-Phe-AFC for up to 1 h. (
We wanted to minimize cost due to continuous cell culturing, as well as assay variation due to cell passage number, by investigating whether this cell assay could be performed with cryopreserved cells. Cellular DPP1 activity was compared using fresh THP-1 and cryopreserved cells ( Table 3 and Fig. 6 ). There was no significant difference in specific DPP1 activity between the different cell preparations, suggesting that cryopreserved cells are just as effective as fresh cells for screening compounds. Although this DPP1 cell assay was constructed in a 96-well format, it could be easily adapted to a 384-well format if a higher compound screening capacity is required.
DPP1 Cell Activity from Fresh and Cryopreserved THP-1 Cells
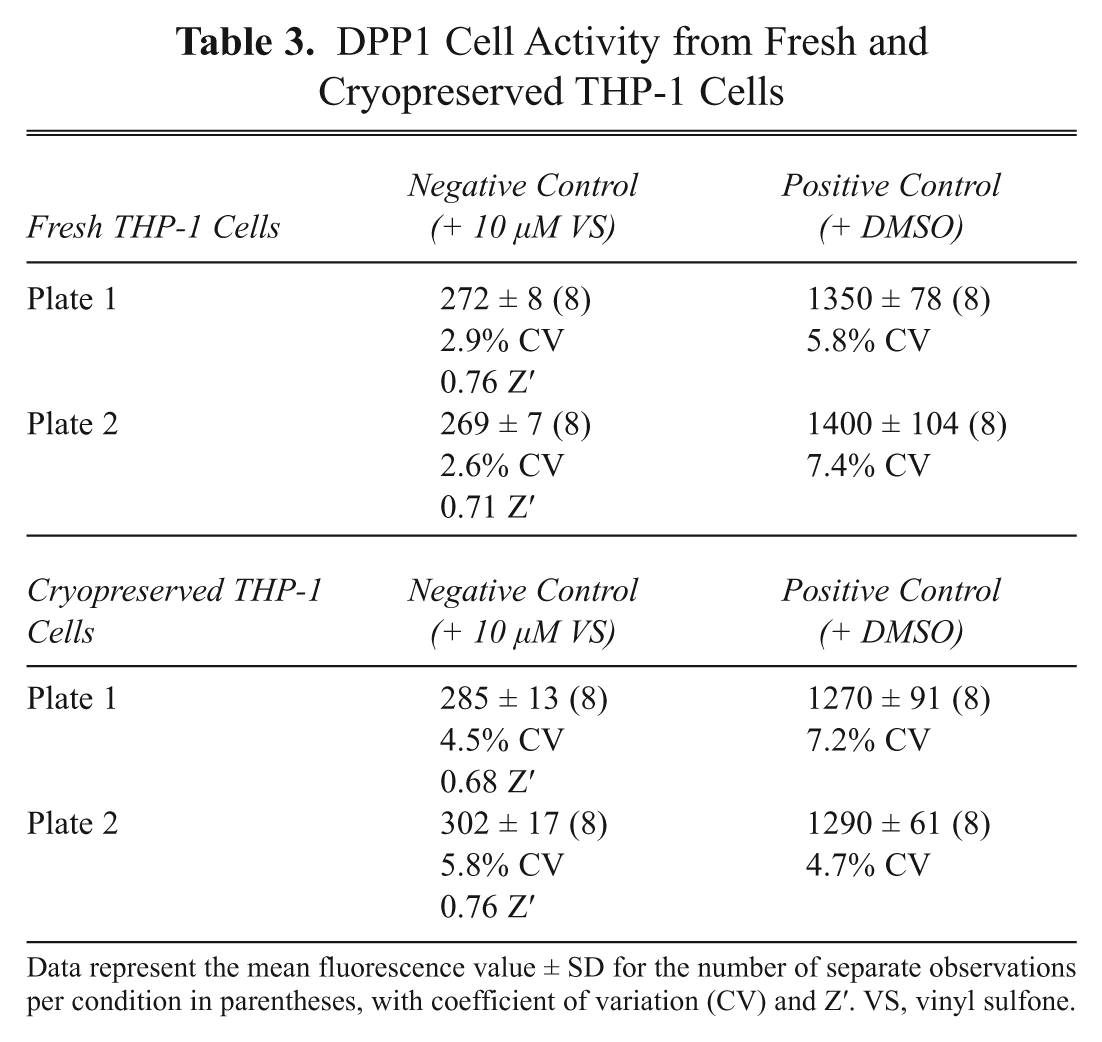
Data represent the mean fluorescence value ± SD for the number of separate observations per condition in parentheses, with coefficient of variation (CV) and Z′. VS, vinyl sulfone.
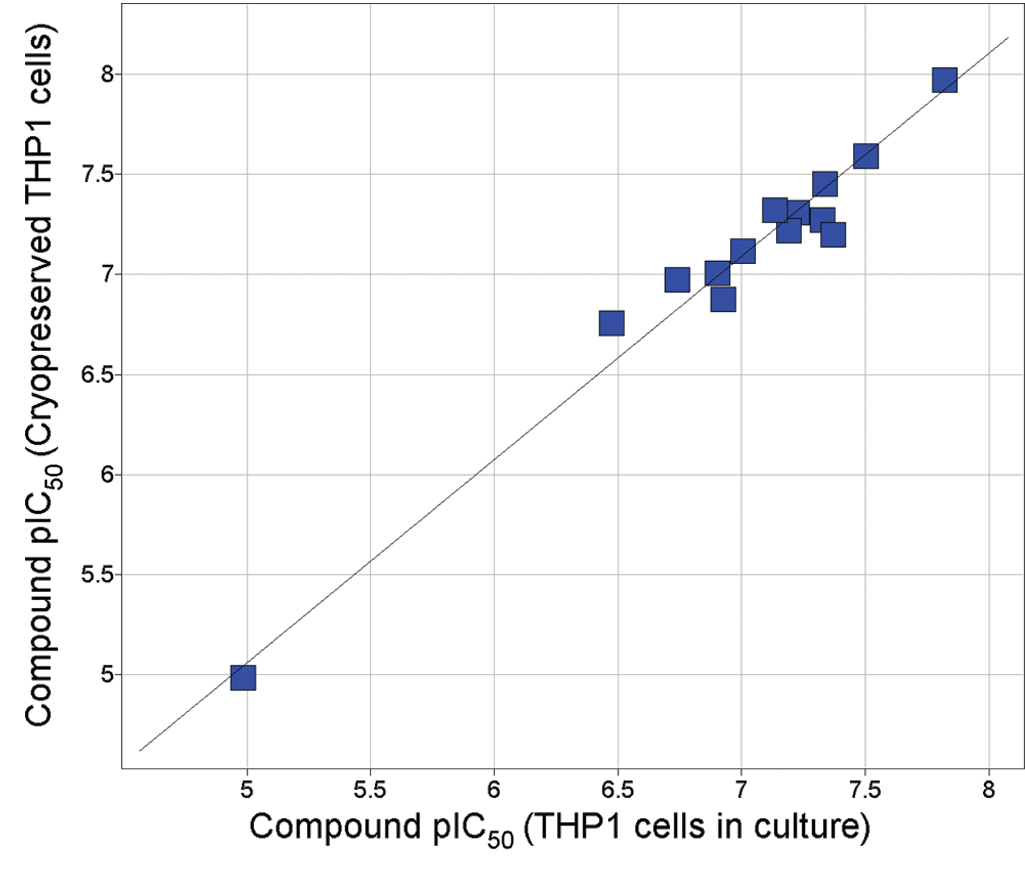
DPP1 cell compound potencies in cryopreserved versus fresh THP-1 cells. Compound pIC50 values were determined as described in Materials and Methods. Data represent the arithmetic mean pIC50 from 2 separate observations per condition for 19 compounds.
In summary, we have identified a suitable substrate (i.e., H-Gly-Phe-AFC) for DPP1 that allowed easy detection of its cellular activity. We validated that DPP1 was responsible for the cellular hydrolysis of this substrate by
Demonstrating inhibition of cell activity by a very selective DPP1 inhibitor (i.e., VS) but not by a range of other protease inhibitors,
Showing good correlation between enzyme inhibition and cell activity inhibition for a range of in-house structurally and physicochemically similar DPP1 inhibitors, and
Describing the localization of this cell activity to lysosomes by using cell imaging to locate DPP1 product formation and inhibition by lysosomal H+-ATPase inhibitor (i.e., bafilomycin A1) and lack of inhibition by the cell-impermeable DPP1 inhibitor (i.e., cystatin).
Finally, the DPP1 cell assay as described above is a simple, dynamic, noninvasive, and high-throughput screen for detecting lysosomal DPP1 activity and inhibition.
Footnotes
Acknowledgements
We thank Mark Furber, Chris Luckhurst, Linda Stein, Sara Dodd, Lorna Ewart, Anna-Karin Tidén, Andy Hargreaves, Chris Roberts, and Andy Walkland for their discussions and constant support.