
Abstract
Functional cell signaling assays have become important tools for measuring ligand-induced receptor activation in cell-based biomolecular screening. Guanosine-5′-triphosphate (GTP) is a generic signaling marker responsible for the first intracellular signaling event of the G-protein-coupled receptors (GPCRs). [35S]GTPγS binding assay is the classical well-established method for measuring agonist-induced G-protein activation requiring a separation of free and bound fractions prior to measurement. Here a novel, separation-free, time-resolved fluorescence GTP binding assay has been developed based on a non–fluorescence resonance energy transfer (FRET) single-label approach and quenching of a nonbound europium-labeled, nonhydrolyzable GTP analog (Eu-GTP). The quenching resonance energy transfer (QRET) method relies on the use of Eu-GTP, providing a time-resolved fluorescent detection as an alternative to the radiolabel [35S]GTPγS assay. Upon activation of recombinant human α2A-adrenoceptors (α2A-AR) expressed in Chinese hamster ovary cells, guanosine-5′-diphosphate is released from the α-subunit of Gi-proteins, enabling the subsequent binding of Eu-GTP. Activation of α2A-AR with 5 different α2-AR agonists was measured quantitatively using the developed QRET GTP assay and compared to [35S]GTPγS and heterogeneous Eu-GTP filtration assays. Equal potencies and efficacy rank orders were observed in all 3 assays but with a lower signal-to-background ratio and increased assay variation in the QRET assay compared to the Eu-GTP filtration and the nonhomogeneous [35S]GTPγS binding assays.
Introduction
C
G-protein-coupled receptors (GPCRs) represent one of the largest protein superfamilies in the human genome. These membrane-bound cell surface receptors mediate the effects of a wide variety of endogenous and exogenous ligands and regulate many important cellular responses. GPCRs are involved in many physiological processes, and their dysfunctions have been associated with various common diseases. The location of GPCRs in the plasma membrane makes them extremely attractive targets for pharmaceuticals (i.e., it is estimated that about 40%-50% of the current clinically used drugs act on GPCRs). 2 The GPCRs closely associate with the G-protein subunits Gα (39-46 kDa) and the 36- to 37-kDa β and 5- to 10-kDa γ subunit (Gβγ) dimer. At agonist activation, a conformational change of a GPCR promotes binding of guanosine-5′-triphosphate (GTP) to the Gα subunit in exchange for guanosine-5′-diphosphate (GDP). 3-6 Guanine nucleotide exchange is an early intracellular event in the signal transduction process, being a generic indicator for signaling to all GPCRs. The radiolabel-based [35S]GTPγS binding assay is widely used to implicate such a signaling process. The [35S]GTPγS binding assay measures the level of agonist-activated GPCR activation by determining the binding of the nonhydrolyzable [35S]GTPγS analog to the G-protein α-subunit. 7,8
Time-resolved fluorometry (TRF) is a well-established alternative technology to radioisotopic assays in many high-throughput applications. 9 The use of TRF has already been described for measuring G-protein activation of, for example, human α2A-adrenergic, 10 cytokine, 11 muscarinic, 12 dopamine, 13,14 opioid, 15 and serotonin 16,17 receptors. Receptor activation was assessed by measuring the binding of a europium-labeled GTP analog (Eu-GTP) to Gα protein on cell membranes. This highly sensitive, nonradioactive functional method potentially improves the screening processes as problems related to waste and storage of radioactive materials can be omitted. However, a separation of bound and nonbound Eu-GTP must still be carried out using filtration or washing. 10
We have explored the use of novel, homogeneous assay formats based on the single time-resolved fluorescence label strategy and a nonspecific quenching mechanism (quenching resonance energy transfer [QRET] technique). The first demonstration of the quenching assay principle shows the potential of the method for receptor-ligand binding studies in drug discovery. 18 The QRET technique is a novel separation-free assay method based on time-resolved fluorescence detection technology using lanthanide(III) chemistry and soluble fluorescence quenchers at the lanthanide(III) emission wavelength. Here we describe the use of the QRET technology for monitoring the activation of GPCRs in a GTP binding assay. We compared the new method with 2 previously established heterogeneous GTP binding assays: [35S]GTPγS and nonradioactive Eu-GTP assays. 7,8,10
Materials and Methods
Materials
[Ethyl-3H]RS-79948-197 (specific activity 81 Ci/mmol) was obtained from GE Healthcare Life Sciences (Buckinghamshire, UK). [35S]GTPγS (specific activity 1250 Ci/mmol) was obtained from PerkinElmer Life and Analytical Sciences (Boston, MA). (−)-Epinephrine, clonidine, (−)-norepinephrine, oxymetazoline, brimonidine, bovine serum albumin (BSA), and 1,1,3,3,3′,3′-hexamethylindodicarbocyanine iodide were purchased from Sigma-Aldrich (St. Louis, MO). Cell culture reagents were supplied by GIBCO® (Invitrogen Life Technologies, Inc., Rockville, MD), and fetal bovine serum was from Autogen Bioclear UK Ltd. (Wiltshire, UK). Other reagents were of analytical or reagent grade and were purchased from commercial suppliers. GDP, MgCl2, NaCl, 50 mM HEPES (pH 7.4), and GTP wash solution were obtained from PerkinElmer, Wallac (Turku, Finland). AcroWell 96-well filter membrane bottom plates (GHP membrane) were from Pall Life Sciences (East Hills, NY), and black 96-well plates were obtained from PerkinElmer. Eu-labeled GTP was guanosine-5′-triphosphate coupled with a fluorescent chelate, 19 obtained from PerkinElmer’s DELFIA® GTP Binding Kit.
Cell culture
A Chinese hamster ovary (CHO) cell line stably expressing the cDNA encoding human α2A-adrenoceptors was produced as described by Pohjanoksa et al. 20 Cells were cultured in α−Minimum Essential Medium (α-MEM) supplemented with 26 mM NaHCO3, 50 U/mL penicillin, 50 µg/mL streptomycin, and 5% heat-inactivated fetal bovine serum supplemented with 200 µg/mL of the neomycin analog G418 (Geneticin®; Sigma-Aldrich). Cells were grown in a humidified incubator at 37°C/5% CO2.
Membrane preparation
Cell membranes were prepared as previously described. 20 Membranes were suspended in 10 mM Tris buffer (pH 7.5) containing 0.1 mM EDTA and aliquoted and stored at −74°C. Protein concentrations were determined according to the method of Bradford 21 using bovine serum albumin as reference.
Saturation binding assay
Receptor density of CHO cells expressing human α2A-adrenoceptor was determined with saturation binding experiments as described previously, 22 using the α2-antagonist radioligand [ethyl-3H]RS-79948-197 (0.021-4 nM). Experiments were performed in triplicate and repeated 3 times. Equilibrium dissociation constant (Kd) and receptor density (Bmax) were calculated from saturation binding data using GraphPad Prism Software (San Diego, CA).
GTP binding assays
A homogeneous GTP binding assay based on the QRET technique was developed and compared to well-known heterogeneous [35S]GTPγS and time-resolved fluorescence filtration GTP binding assays. EC50, Emax, and signal-to-background ratios (the signal obtained in the presence of an agonist concentration causing a maximal response/the background signal from the system containing no agonist) were calculated in the comparative tests.
Heterogeneous [35S]GTPγS binding assay
Agonist-induced stimulation of [35S]GTPγS binding was measured essentially as described previously. 23 Briefly, membranes were thawed and resuspended in incubation buffer (50 mM Tris, 1 mM EDTA, 5 mM MgCl2, 20 mM NaCl, 1 µM GDP, 1 mM dithiothreitol [DTT], 30 µM ascorbic acid, pH 7.4). All incubations were performed on 96-well Millipore MultiScreen MAFB/MSFB glass fiber filter plates (Millipore Corp., Bedford, MA). Samples containing 5 µg of membrane protein were incubated with agonists and 0.1 nM [35S]-GTPγS for 30 min at 37°C. Reactions were terminated by vacuum filtration using a Millipore MultiScreen Vacuum Manifold. The filter plates were washed 3 times with ice-cold wash buffer (20 mM Tris, 1 mM EDTA, 5 mM MgCl2, pH 7.4). Filters were dried, and 50 µL SuperMix scintillation cocktail (PerkinElmer, Wallac) was added into each well. The incorporated radioactivity was measured using a MicroBeta scintillation counter (PerkinElmer, Wallac). All assays were performed in duplicate. Additional [35S]GTPγS control experiments were performed with the soluble quencher (1,1,3,3,3′,3′-hexamethylindodicarbocyanine iodide) that was used in the homogeneous QRET assay to test whether the quencher affected the binding properties of [35S]GTPγS in the assay. The quencher (2.3 µM) was added upon completion of the [35S]GTPγS incubation step and 5 min before vacuum filtration. This simulated the procedure of the homogeneous QRET assay.
Heterogeneous time-resolved fluorescence assay
The Eu-GTP binding assay was performed in AcroWell filter plates using a 2-step incubation protocol. The reaction was started by adding an aliquot of the membrane suspension (10 µg protein) to reaction buffer containing 50 mM HEPES buffer, pH 7.4, supplemented with 50 mM NaCl, 5 mM MgCl2, 2.5 µM GDP, and varying concentrations of the following agonists to a total volume of 100 µL: (−)-epinephrine, clonidine, (−)-norepinephrine, oxymetazoline, and brimonidine. After a 30-min preincubation on a plate shaker (slow shaking), 10 nM europium-labeled GTP was added. Incubation was continued for another 30 min, and the reaction was terminated by vacuum filtration (MultiScreen Vacuum Manifold, Millipore), followed by 2 washes with ice-cold buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl2). The bound Eu-GTP was measured immediately after filtration with a Victor 2 fluorescent plate reader (PerkinElmer, Wallac). The delay and integration times were 400 µs, and the excitation and emission wavelengths were 340 and 615 nm, respectively.
Homogeneous QRET assay
CHO cell membranes (30 µg protein) were dispensed into 96-well plates in 50 mM HEPES, pH 7.4. Agonists were added at varying concentrations to a total volume of 100 µL in the reaction buffer (50 mM HEPES, pH 7.4; 2.5 µM GDP; 5 mM MgCl2; 50 mM NaCl; 0.001% BSA) and incubated for 30 min at room temperature in the dark with slow shaking. Thereafter, 10 µL of Eu-GTP (final concentration 10 nM) was added, and the reaction mixture was incubated for another 30 min under the same conditions. After incubation, 20 µL of soluble quencher (1,1,3,3,3′,3′-hexamethylindodicarbocyanine iodide) at 15 µM concentration (final concentration 2.3 µM) was added, the plate was shaken, and time-gated fluorescence was measured with a Victor 2 fluorescent plate reader. The delay and integration times were 400 µs, and the excitation and emission wavelengths were 340 and 615 nm, respectively.
Results and Discussion
Saturation binding assay
The receptor density (Bmax) determined with 3 independent saturation binding experiments was 3800 ± 60 fmol/mg of protein for CHO cell membranes stably expressing human α2A-AR. The equilibrium dissociation constant (Kd) of the radioligand [ethyl-3H]RS-79948-197 was 0.11 ± 0.01 nM.
Homogeneous QRET assay
A simple homogeneous nonradioactive QRET assay for GTP binding was developed using a single-label approach and quenching of time-resolved luminescence in solution ( Fig. 1 ). With agonist-induced activation of α2A-AR in CHO cell membranes and subsequent binding of Eu-labeled GTP to Gαi protein, the fluorescence of Eu-GTP was protected and measured at 615 nm. When no receptor activation was carried out, Eu-GTP remained in solution, and nonbound Eu-GTP was quenched in solution using 1,1,3,3,3′,3′-hexamethylindodicarbocyanine iodide at 2.3 µM quencher concentration. The developed assay was performed on 96-well plates to compare the results with previously validated [35S]GTPγS and Eu-GTP 96-plate filtrations assays. The QRET assay was conducted in 2 steps: 30-min agonist preincubation and 30-min incubation with Eu-GTP, as well as subsequent addition of the quencher and detection of the time-gated Eu(III) luminescence signal.
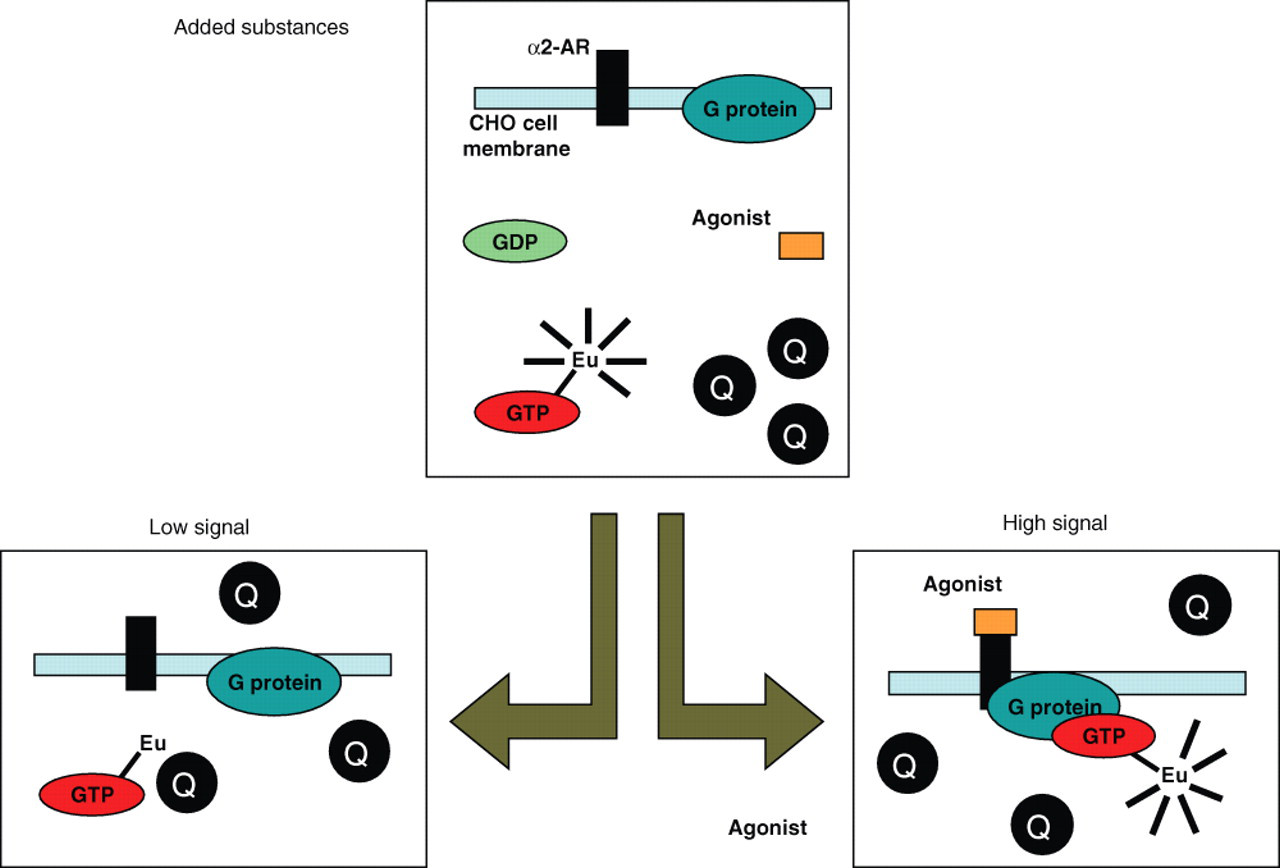
The principle of the homogeneous Eu-GTP binding assay using the quenching resonance energy transfer (QRET) principle.
The assay conditions of the homogeneous QRET assay were chosen based on our previous optimized assay conditions. 10 However, the composition of the reaction buffer was modified by adding 0.001% BSA to reduce any possible nonspecific binding. In the employed [35S]GTPγS binding assay, the concentrations of GDP and NaCl markedly affect the stimulation efficacy and signal-to-background ratio of [35S]GTPγS binding. High concentrations of GDP (10 µM) or NaCl (150 mM) effectively decrease the stimulation efficacy of partial agonists. 10 Therefore, in the present study, the GDP and NaCl concentrations were chosen not to exceed these concentrations.
For the homogeneous QRET assay, the quantity of membrane protein in the sample was optimized, varying from 10 to 60 µg per well in norepinephrine-induced stimulations ( Fig. 2 ). Agonist-stimulated binding of Eu-GTP was detectable already at 10 µg membrane protein, but the signal-to-background ratio was insufficient. With 30 µg membrane protein, the signal-to-background ratio was 2, reaching an apparent saturation. Thus, 30 µg membrane was chosen for further experiments.
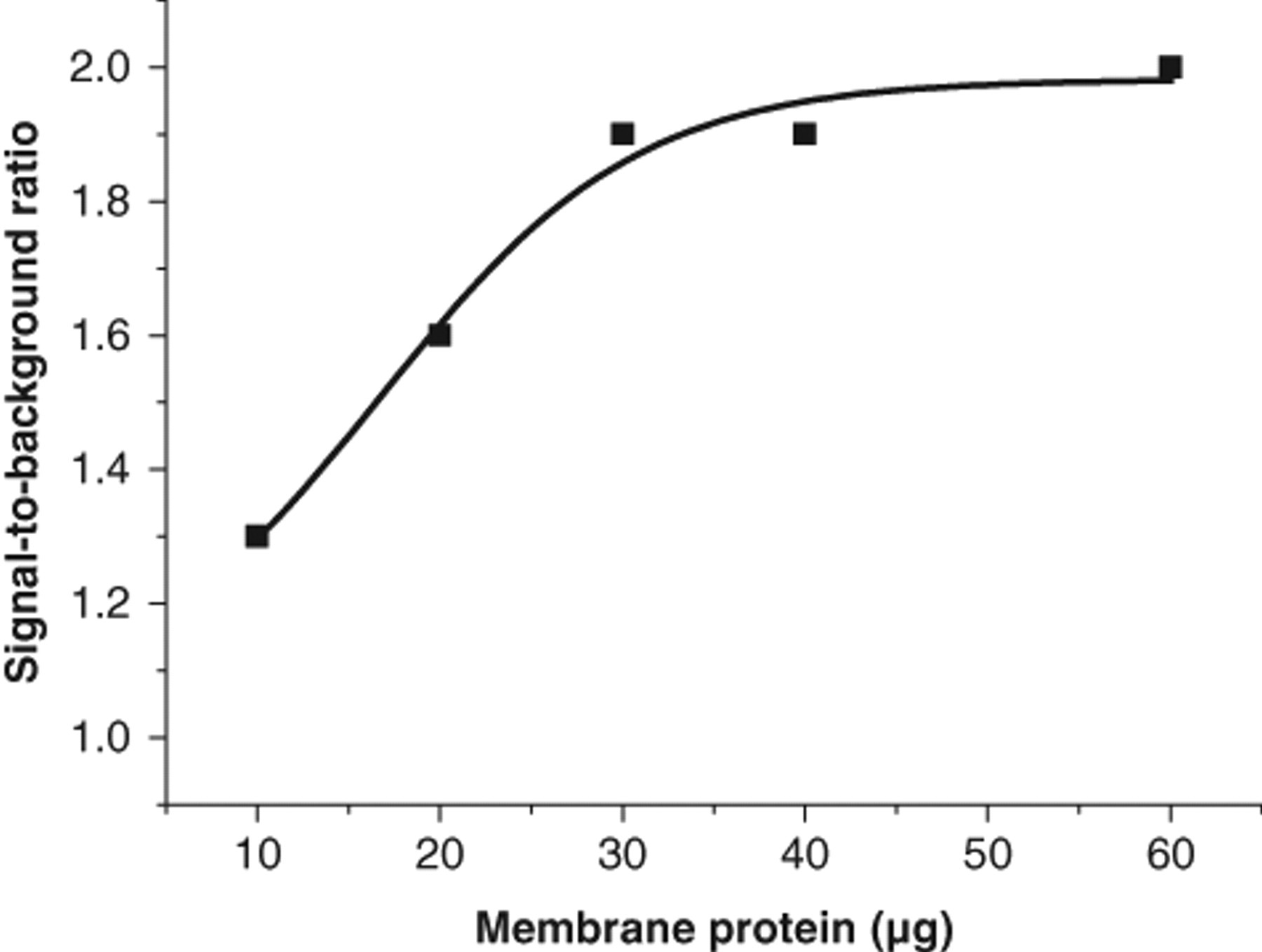
Optimization of the membrane protein concentration in the homogeneous quenching resonance energy transfer (QRET) assay.
Dose-response experiments were carried out under the optimized assay conditions for 5 agonists that are full or partial agonists at α2A-AR: (−)-norepinephrine, (−)-epinephrine, brimonidine, clonidine, and oxymetazoline ( Fig. 3a ). EC50 and Emax values were calculated from the dose-response data and are given in Table 1 . The signal-to-background ratio for (−)-norepinephrine was 1.9, and the average coefficient of variation was 13.1% for the different agonists. The quencher was found to have an insignificant effect on assay performance, as studied with the radioligand [35S]GTPγS assay (data not shown).
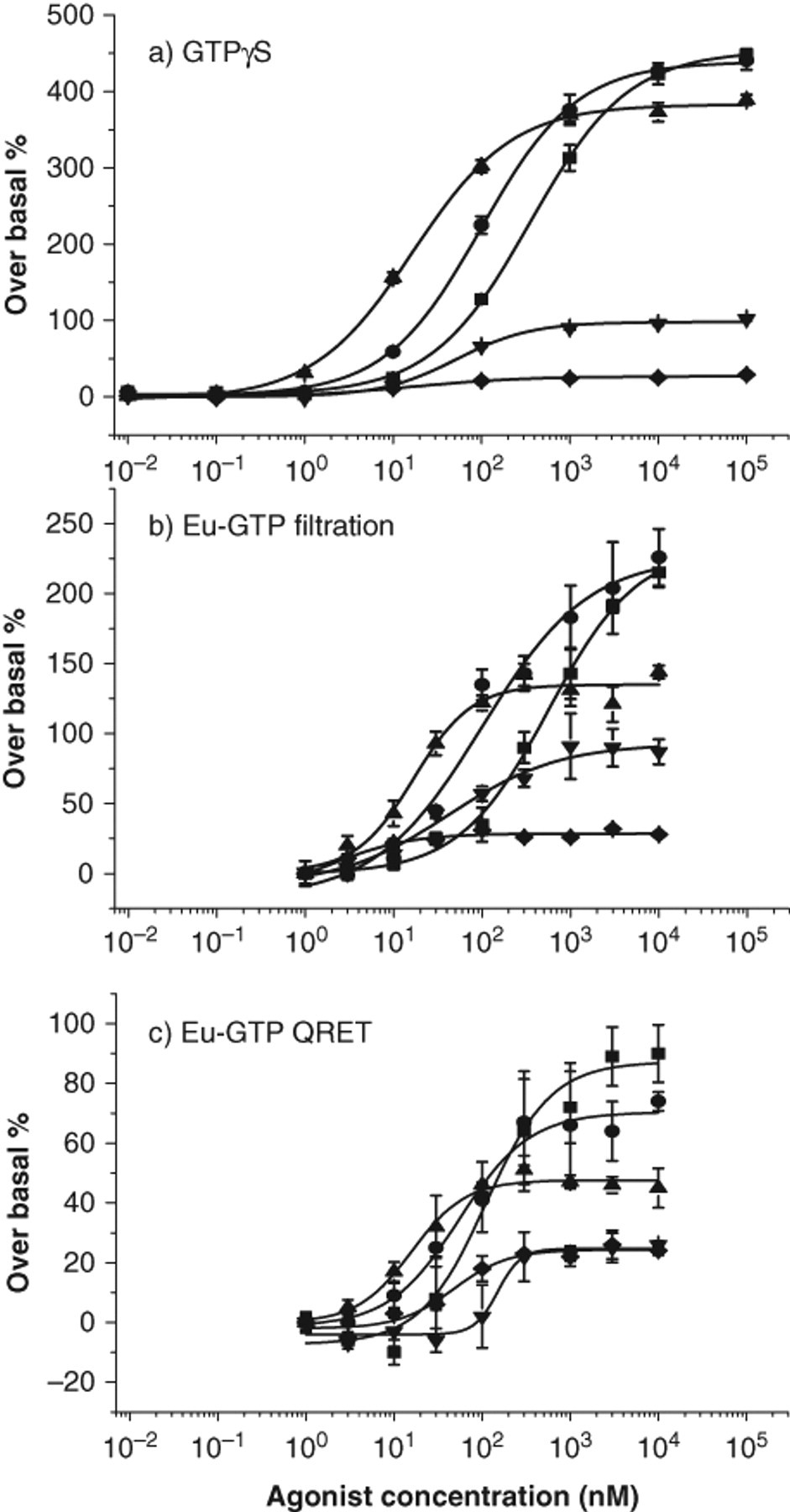
Dose-response curves of guanosine-5′-triphosphate (GTP) binding assays:
(
Characterization of the GTP Binding Assays
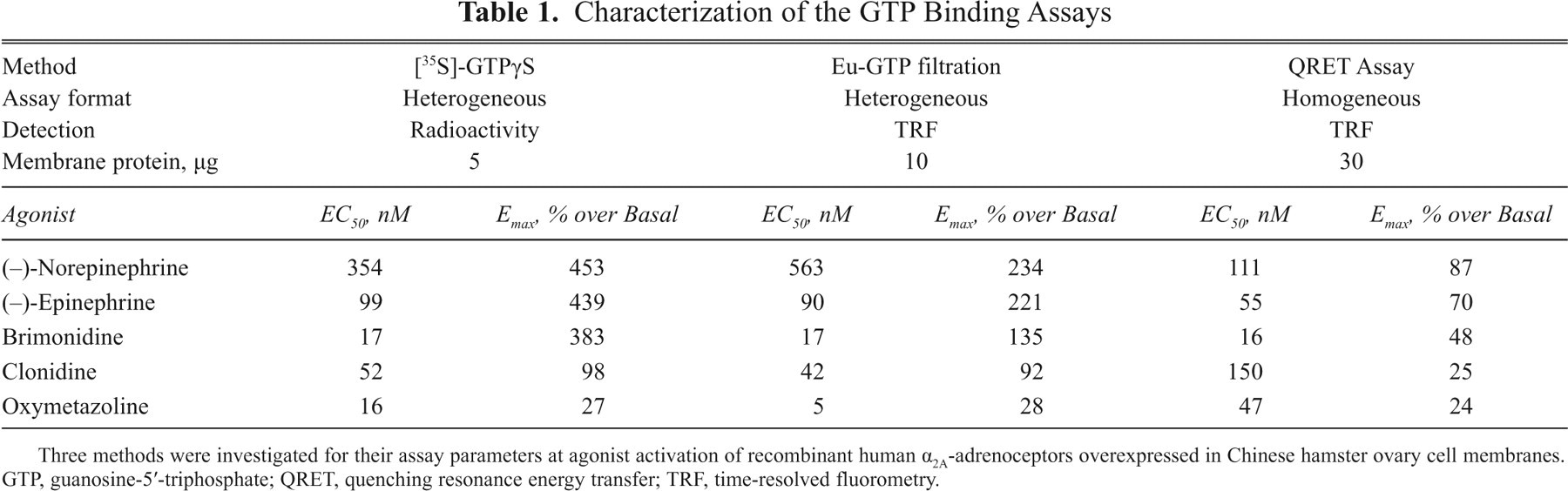
Three methods were investigated for their assay parameters at agonist activation of recombinant human α2A-adrenoceptors overexpressed in Chinese hamster ovary cell membranes. GTP, guanosine-5′-triphosphate; QRET, quenching resonance energy transfer; TRF, time-resolved fluorometry.
After agonist exposure, a significant difference between the basal and agonist-induced binding was observed for all tested agonists. The full agonists resulted in a higher efficacy compared with the partial agonists showing a reduced basal binding activity. Brimonidine has been reported to have a lower efficacy than (−)-norepinephrine and (−)-epinephrine, as observed here too.
Heterogeneous time-resolved fluorescence assay
Heterogeneous nonradioactive assays were performed using the commercially available test kit. 10 The reaction conditions were the same as in the new homogeneous assay system, excluding BSA and using only 10 µg membrane protein. After the 2-step incubation, nonbound Eu-GTP was separated using filtration on a vacuum manifold.
The full and partial agonists were investigated in this heterogeneous assay format, and the dose-response curves are shown in Figure 3b . Generally, in comparison to the homogeneous assay format, similar rank orders of efficacy and potency were observed. However, the maximal binding efficacies (Emax) of the full agonist were higher than in the homogeneous assay format ( Table 1 ). The average coefficient of variation was 8.6% for the different agonists, and the signal-to-background ratio of 3.3 was obtained for (−)-norepinephrine.
Heterogeneous [35S]GTPγS binding assay
Radiolabel binding assays are well established in biomolecular screening laboratories; therefore, an in-house developed [35S]GTPγS binding assay was chosen for comparison. 23 The assay conditions differed from the nonradioactive binding assays as the buffer constitution was different (Tris vs. HEPES), and only 5 µg of membrane protein was used. After incubation, the reaction mixture was vacuum filtered through a glass fiber filter to separate nonbound [35S]GTPγS from the bound fraction.
For the radiolabeled-based [35S]GTPγS binding assay, previously similar high efficacy has been reported for the full agonists (−)-norepinephrine, (−)-epinephrine, and brimonidine, as observed in the present study. 10 The signal-to-background ratio for (−)-norepinephrine was 5.5, which was clearly higher than that for the nonradioactive methods. Of the tested methods, the [35S]GTPγS binding assay is by far the most established method, resulting in the lowest coefficient of variation, 1.9%.
Comparison of the methods
The comparison of the methods was based on the characteristics of the dose-response data. The evaluation of the half-maximal effective concentration EC50 and the maximal binding efficacy Emax is given in Table 1 . The EC50 values of 2 endogenous agonists—(−)-norepinephrine and (−)-epinephrine—as determined in the QRET homogeneous Eu-GTP binding assay were somewhat lower than the corresponding values in the radiolabel and heterogeneous, nonradioactive binding assays. The EC50 value of brimonidine was the same independently of the method used. The EC50 values of clonidine and oxymetazoline may not be directly comparable as the signal-to-background ratios of the partial agonists were generally weak, and relatively high assay variation was observed.
The rank orders of agonist efficacy were very similar in all assay formats. However, differences in the Emax values and signal-to-background ratios were measured ( Table 1 ). The well-established [35S]GTPγS assay resulted in the highest signal-to-background ratio and the homogeneous Eu-GTP the lowest. This suggests that the Eu-GTP binding may potentially be weaker than that of the radiolabeled GTP. The concentration of membrane protein required per assay was 2-fold higher for the heterogeneous and 6-fold for the homogeneous Eu-GTP assay than that of the GTPγS assay, which also partially supports the weaker binding property of Eu-GTP.
The assay variation was very low in the [35S]GTPγS assay. The [35S]GTPγS was well established in our hands in comparison with the TRF Eu-GTP filtration assay. The TRF Eu-GTP filtration assay was carried out in the Laboratory of Biophysics during this study for the first time, which may have resulted in the higher variation of the Eu-GTP assay. Evidently, more work needs to be carried out to improve the reproducibility of the QRET Eu-GTP assay, and automation of the manual Eu-GTP assay may have to be established.
Conclusions
A homogeneous assay based on the QRET technique was developed for the GTP binding assay. The developed assay was compared with the heterogeneous [35S]GTPγS and Eu-GTP binding assays at stimulation of α2-AR on CHO cell membranes with comparable efficacy rank order and EC50 values. The [35S]GTPγS assay resulted in the lowest assay variation and the highest signal-to-background ratio of the methods. Although further fine-tuning of the assay parameters, possibly through new synthetic strategies of the Eu-labeled GTP, is required in the homogeneous QRET method, this study shows the potential of the single-label time-resolved fluorescence concept in assaying old targets with a novel means. As there is no need to label the target receptor in the QRET technique, this additional free dimension allows the development of new nonradioactive technologies for drug discovery purposes.
Footnotes
Acknowledgements
The work was supported by the Finnish Funding Agency for Technology and Innovation, Tekes.