
Abstract
Background:
Rivaroxaban, a direct factor Xa inhibitor, has been developed to meet clinical needs in a broad range of indications in adults: prevention of venous thromboembolism after elective hip or knee replacement surgery, treatment and secondary prevention of venous thromboembolism, prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation having one or more risk factors, and in Europe, prevention of atherothrombotic events after an acute coronary syndrome in patients with elevated cardiac biomarkers. However, the precise dose and regimen vary with the indication, leading to this effort to provide clarity concerning the appropriate use of rivaroxaban. This article reviews the clinical development program for rivaroxaban and summarizes the evidence for each approved, indication-specific dose regimen.
Results:
Although initially investigated for twice-daily dosing, early observations, including the finding that the pharmacodynamic effects of rivaroxaban last longer than the elimination half-life, suggested that once-daily dosing might be attainable and effective. These observations were evaluated within the extensive phase II program, which, together with pharmacology studies, provides the evidence underpinning the selection of once-daily regimens for most, but not all, of the approved clinical indications for rivaroxaban.
Conclusion:
The evidence for each dosing regimen demonstrates that although pharmacology studies are of paramount importance, dose regimens must be subjected to careful empirical validation. Once-daily dosing was shown to be clinically appropriate for most rivaroxaban indications. Furthermore, a “one size fits all” approach to dosing frequency is unlikely to result in a regimen that yields optimal patient outcomes across different indications.
Introduction
In 2008, rivaroxaban became the first novel oral direct inhibitor of activated factor X (Xa) to receive marketing authorization for a clinical indication, namely, the prevention of venous thromboembolism (VTE) in adult patients undergoing elective hip or knee replacement surgery. Since then, an extensive clinical development program has resulted in regulatory approvals for several further clinical indications in adults: the treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE) and the prevention of recurrent DVT/PE, the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation (AF) having one or more risk factors, and in Europe, the prevention of atherothrombotic events after an acute coronary syndrome (ACS) in patients with elevated cardiac biomarkers. 1 However, each of these therapeutic indications has a specific regimen involving different dosing, administration frequencies, and treatment durations (Table 1). Understanding the rationale behind the determination of each regimen, particularly why once-daily (OD) dosing is deemed appropriate for most clinical indications and why twice-daily (BID) dosing is recommended for others, will help practitioners to avoid confusion in selecting the optimal dosing regimen for each patient.
Dose Regimens in Approved Rivaroxaban Indications. 1

Abbreviations: ACS, acute coronary syndrome; ASA, acetylsalicylic acid; AF, atrial fibrillation; BID, twice daily; CrCl, creatinine clearance; DVT, deep vein thrombosis; OD, once daily; PE, pulmonary embolism; THR, total hip replacement; TKR, total knee replacement; VTE, venous thromboembolism.
aStandard dose for continued treatment and secondary prevention. Dose reduction to 15 mg OD may be considered if the patient’s risk of bleeding outweighs their risk of recurrent VTE.
bPotentially lifelong.
cStandard dose for patients with normal renal function or mild renal impairment (CrCl ≥ 50 mL/min). Dose should be reduced to 15 mg OD in patients with mild/moderate renal impairment (CrCl 15-49 mL/min).
dNot presently approved in the United States. European approval is for the prevention of atherothrombotic events after ACS in patients with elevated cardiac biomarkers.
eAdministered in combination with ASA or ASA plus clopidogrel or ticlopidine.
The pharmacokinetic properties of rivaroxaban might ordinarily suggest a BID dosing regimen. However, certain early observations (discussed subsequently) suggested that OD dosing might be achievable. Other authors have noted that although clinical pharmacology studies can and should provide guidance, empirical clinical validation of dosing regimens in the relevant patient population must be the definitive arbiter for optimization. 2
This article will review the early characterization and clinical development of rivaroxaban, including the initial observations suggesting that OD dosing might be feasible. The subsequent step-by-step clinical validation program for each indication will then be described. The article will also illustrate how the choice of OD or BID dosing was based on solid evidence derived from a large-scale, comprehensive phase II dose-selection program. In doing so, the point will be made that for a multi-indication drug, a “one size fits all” approach does not necessarily yield the ideal outcome in every indication, and thus, dosing regimens should always be optimized for the various pathophysiologies encountered.
The Early Studies
The preclinical characterization of rivaroxaban is briefly described in section 1 of the Supplementary Material. These preclinical observations were followed by phase I studies in healthy humans. Single-dose applications of up to 80 mg and multiple-dose applications of up to 30 mg BID showed dose-dependent pharmacokinetics and inhibition of factor Xa, as well as prolongation of prothrombin time, with close correlation between the respective pharmacodynamic parameters and the plasma concentrations. Maximal plasma drug concentration (Cmax) and maximal effects on pharmacodynamics tests were reached 2.5 to 4 hours after rivaroxaban intake. 3,4 Rivaroxaban was eliminated with a terminal half-life (t1/2) of approximately 5 to 9 hours from the steady state, irrespective of dose, in young, healthy volunteers, 4 suggesting the feasibility of a BID dosing regimen. Conversely, in elderly patients, the half-life was prolonged to 11 to 13 hours, which is more in line with OD dosing. 5,6 In addition, consistent with the mode of action, a prolonged influence of rivaroxaban on the inhibition of the endogenous thrombin potential (ETP), a measure of thrombin generation capacity in platelet-rich plasma, was observed after activation by either collagen or tissue factor. 7,8 The collagen-induced ETP peak was reduced by ∼80% after a 5-mg dose of rivaroxaban and by ∼90% after a 30-mg dose of rivaroxaban, and similar effects on the ETP peak were observed after activation with tissue factor. At 24 hours after the 30-mg dose, inhibition was still ∼40% after collagen activation and ∼20% after tissue factor activation (Figure 1). However, no inhibition of ETP was apparent at 24 hours after a 5-mg dose. 7,8
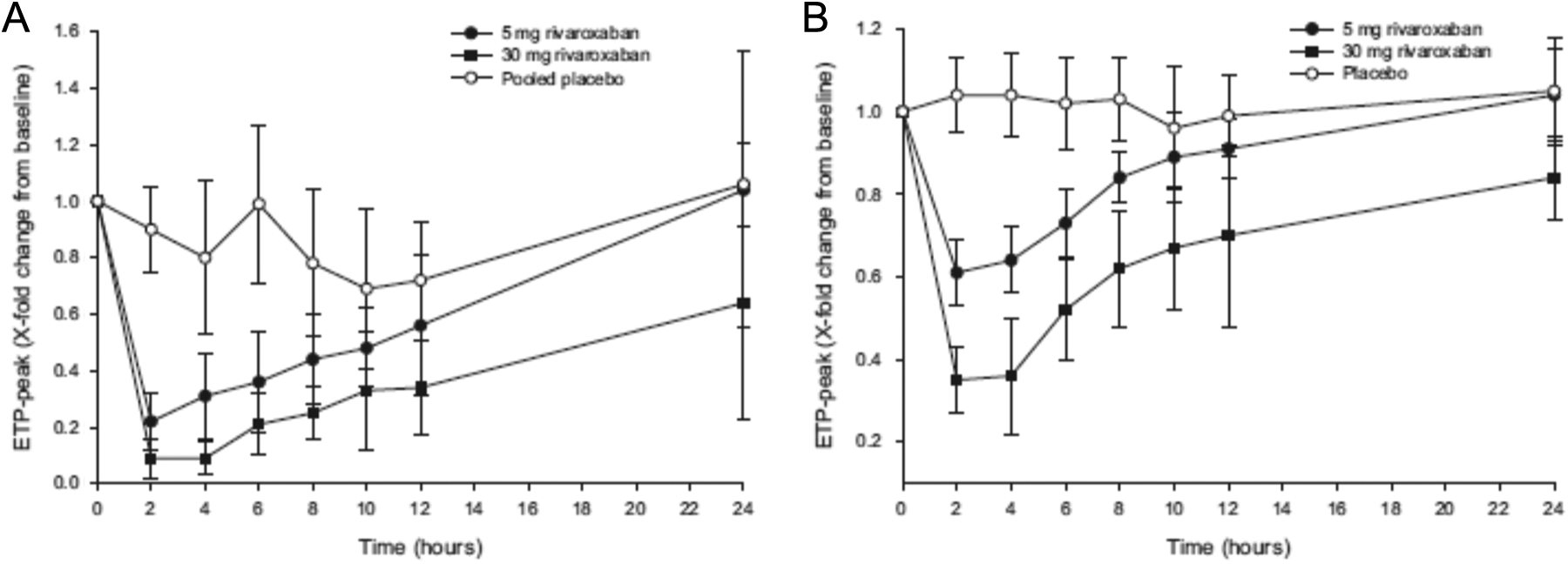
Effect of rivaroxaban on endogenous thrombin potential. 7,8 Effect of rivaroxaban 5 and 30 mg and placebo on (A) ETP peak after activation by collagen (5 μg/mL) and (B) ETP peak after activation by tissue factor (1.4 nmol). Data are mean relative change in baseline ± standard deviation. ETP indicates endogenous thrombin potential.
The area under the concentration–time curve (AUC) correlated inversely with creatinine clearance (CrCl), and CrCl decreased with age. 5,6,9 However, the effect of renal dysfunction on rivaroxaban clearance is moderate, even in patients with severe renal impairment. Notably, despite increases in exposure of approximately 40% to 60% in patients with mild to severe renal impairment, t1/2 was only slightly prolonged, limiting the risk of unexpected accumulation in these patients. 9 Importantly, the drug was safe and well tolerated in all phase I studies, with no apparent increase in the risk of bleeding 3 or clinically relevant increases in bleeding time. 4
Initial Clinical Development Suggesting Feasibility of OD Dosing
Studies suggesting greater patient compliance or adherence with OD therapy versus more frequent dosing schedules have been available for more than 2 decades. 10 Subsequent studies have reported similar findings with respect to cardiovascular medications, 11,12 although it seems probable that the findings are applicable to a wide range of oral medications prescribed for chronic conditions. 13 These findings are important because poor adherence with treatment is associated with poorer outcomes, including increased mortality 11 –13 and increased costs. 13,14 Furthermore, many patients with chronic cardiovascular conditions are taking many different medications at the same time (polypharmacy). Increased medication and refilling complexity (eg, number of prescribers, separate pharmacy visits, etc) are also associated with poorer adherence, 15 suggesting that simplifying drug regimens by reducing dose frequency is likely to be beneficial. 11 Collectively, these observations indicate that compared with more frequent dosing regimens, OD dosing is associated with better adherence to therapy and results in improved outcomes for the treated population. Therefore, it was considered desirable to evaluate the feasibility of OD dosing for rivaroxaban.
The prevention of VTE after elective total hip replacement (THR) or total knee replacement (TKR) is a short-term indication because patients only require postoperative anticoagulation for 2 to 5 weeks after surgery, consistent with the time frame of traumatically induced (transient) hypercoagulability. This, together with the relatively high rate of DVT and the ability to quantify bleeding and adverse events, made VTE prevention after THR/TKR an excellent in vivo model for the initial clinical evaluation of a new anticoagulant. 16
A proof-of-principle, open-label, dose-escalation phase IIa study (ODIXa-HIP1) was conducted in patients undergoing THR. 17 Patients were randomized to rivaroxaban doses of 2.5, 5, 10, 20, or 30 mg BID, commencing 6 to 8 hours after surgery, or to the standard of care (SOC) comparator, 40 mg OD enoxaparin administered subcutaneously and commencing the evening before surgery. Each rivaroxaban dose stage was completed before the next higher dose was initiated. During the course of this study, 2 observations suggesting that OD dosing of rivaroxaban might be feasible were noted. First, a phase I study of the pharmacokinetic/pharmacodynamic interactions of rivaroxaban and enoxaparin showed that both drugs had a similar rapid onset of anti-factor Xa activity (time to Cmax of 2-4 hours), with a nearly identical time course (Figure 2). 18 Because enoxaparin administered OD had been the SOC comparator in the ODXIa-HIP1 trial, this close similarity in the pharmacodynamic time course of rivaroxaban and enoxaparin suggested that OD rivaroxaban might be a viable regimen, especially because anti-factor Xa activity is the leading anticoagulant effect for low-molecular-weight heparins (LMWHs) such as enoxaparin. 19 Second, in another phase I study, rivaroxaban-induced inhibition of factor Xa activity and reduction in ETP were still evident at least 24 hours after administration of doses approximating therapeutic concentrations. 3,7,8 As noted previously, reduction in ETP was evident 24 hours after a 30-mg dose of rivaroxaban but not after a 5-mg dose (Figure 1). 7,8 On the basis of these observations suggesting that OD dosing might be feasible, a further rivaroxaban dose group, 30 mg OD, was added to this study. Treatment continued until mandatory venography 5 to 9 days after surgery. The primary efficacy end point (composite of total DVT, PE, and all-cause mortality) occurred in 10.2% to 23.8% of patients receiving rivaroxaban and in 16.8% of those receiving enoxaparin (Supplementary Table 1), and the dose–response for rivaroxaban was not statistically significant. However, statistically significant dose responses were seen for major VTE (composite of proximal DVT, PE, and VTE-related death) 17 and for major postoperative bleeding (Supplementary Table 2 and Figure 3). 17 Although the trial did not study a 15-mg BID dosing regimen, it is apparent from the dose–response curves that the incidence results for the efficacy and safety outcomes of the 30-mg OD dose were similar to those expected for a 15-mg BID dose (Figure 3). 17 These results provided the first proof-of-principle for the potential clinical use of rivaroxaban as an antithrombotic drug and the first clinical indication that OD dosing might indeed be achievable.
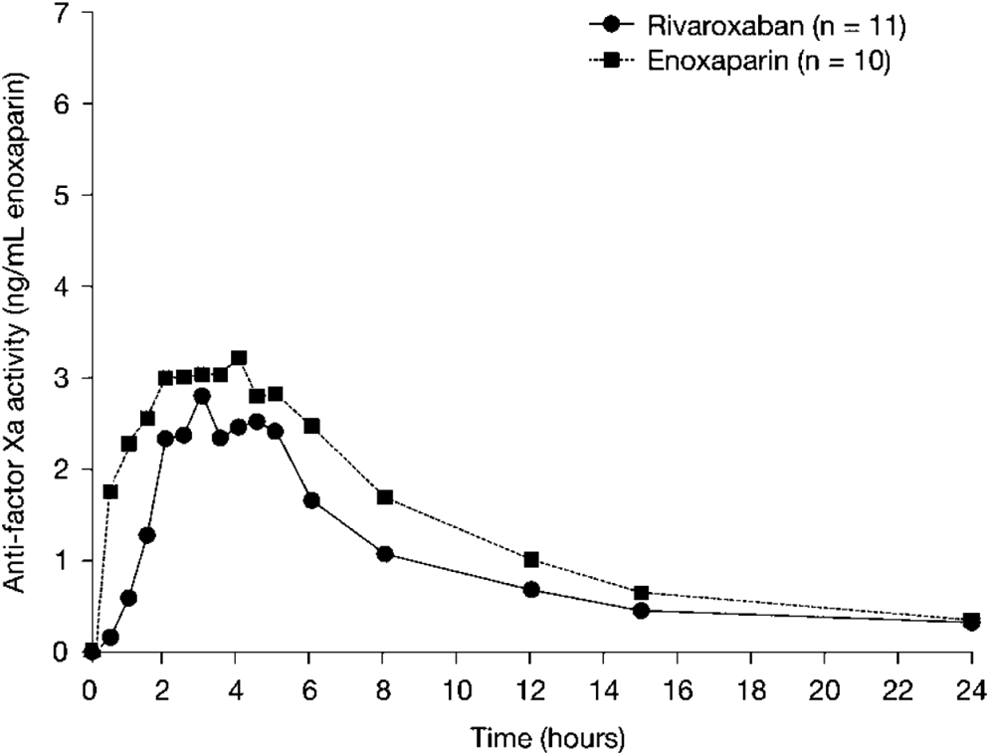
Anti-factor Xa activity of oral rivaroxaban and subcutaneous enoxaparin. 18 Anti-factor Xa activity of oral rivaroxaban 10 mg or subcutaneous enoxaparin 40 mg in healthy male subjects (n = 10-11). Values are medians.
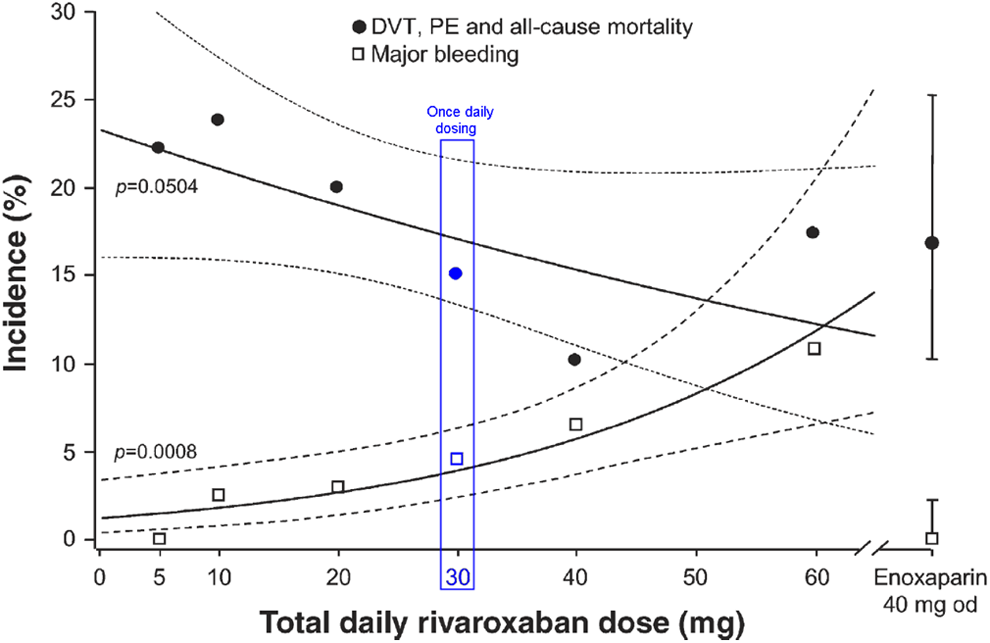
Dose–response relationships for VTE prevention after hip replacement surgery with rivaroxaban for the primary efficacy end point of any DVT, PE, and all-cause mortality (per-protocol population, n = 466) and major, postoperative bleeding (safety population, n = 625). 17 The solid lines are the dose–response curves for rivaroxaban, estimated using logistic regression with dosage as a covariate. The dashed lines represent pointwise 95% confidence intervals. All daily doses were divided and administered BID, except for the 30 mg dose, which was OD. BID indicates twice daily; DVT, deep vein thrombosis; OD, once daily; PE, pulmonary embolism; VTE, venous thromboembolism.
Three further phase II dose-ranging studies were also conducted, all of which used the same protocols and study end points as previously, making this an extensive phase II program with a total of 2866 patients enrolled. Two of these studies, ODIXa-HIP2 20 and ODIXa-KNEE, 21 evaluated BID regimens and are summarized in Supplementary Material (section 2). The conclusion from these 2 studies was that the optimal BID dose of rivaroxaban was 2.5 to 10 mg BID for the prevention of post-THR/TKR VTE, that is, this dose demonstrated potential efficacy with acceptable safety. 20,21 In light of these observations, and the suggestion that OD dosing might be feasible, a fourth phase II dose-ranging study was undertaken to assess OD dosing. This study, ODIXa-OD-HIP, randomized patients to rivaroxaban 5, 10, 20, 30, or 40 mg OD or enoxaparin 40 mg OD. 22 Primary efficacy end point (composite of any DVT, symptomatic confirmed PE, or all-cause mortality) events occurred at a lower rate for all rivaroxaban doses than with enoxaparin, without a significant dose–response (Supplementary Table 1). 22 However, a significant dose–response was observed with major bleeding (Supplementary Table 2). 22 The rate of major venous thromboembolic events with rivaroxaban was similar to that observed for enoxaparin for all rivaroxaban doses, except for the higher rate observed with the lowest dose, 5 mg OD. Furthermore, rates of major postoperative bleeding in this study were similar to those observed with BID dosing in the previous studies, indicating that OD dosing was not associated with an increase in major bleeding. 22 Dose responses for BID and OD dosing are illustrated in Figure 4. The results of this study provided strong evidence in support of OD dosing and led to the selection of rivaroxaban 10 mg OD as the dose regimen to be evaluated in the comprehensive phase III RECORD (REgulation of Coagulation in ORthopaedic surgery to prevent Deep vein thrombosis and pulmonary embolism) program.
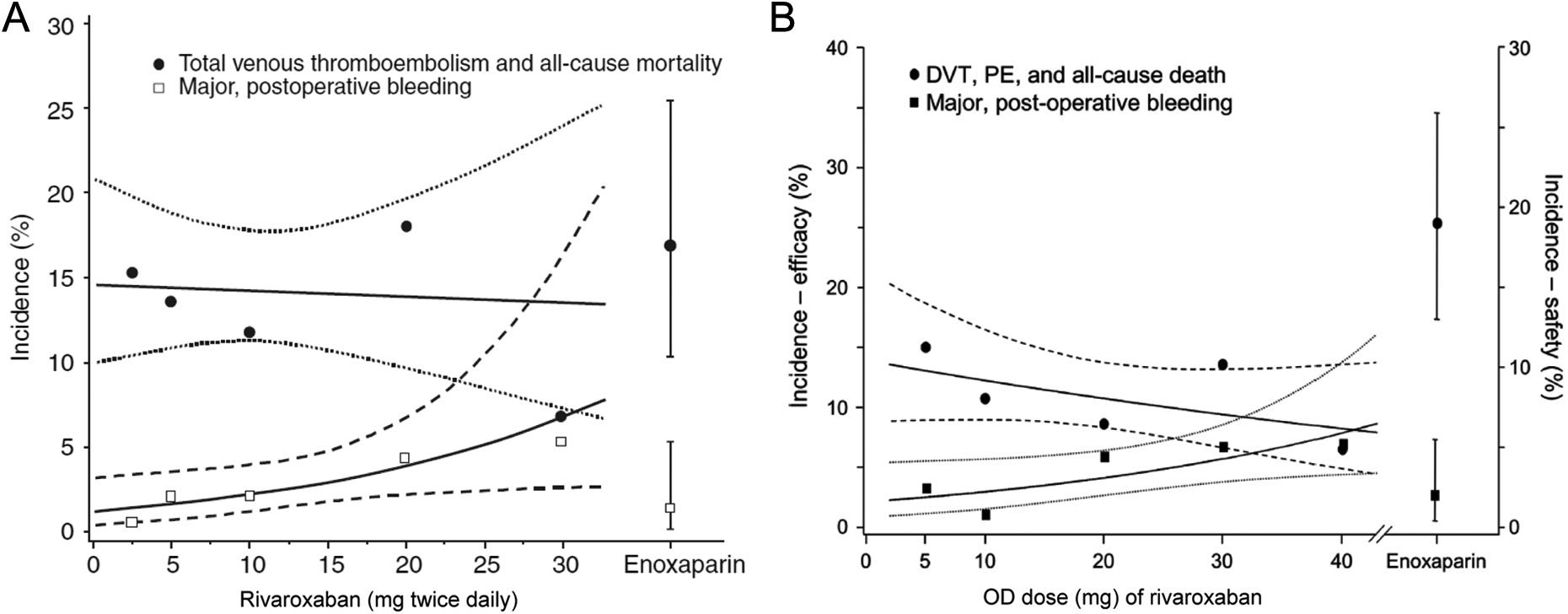
Dose–response curves for venous thromboembolism prevention after hip replacement surgery by rivaroxaban administered (A) BID or (B) OD. 20,22 A, Dose–response relationship between BID rivaroxaban and the primary efficacy end point (DVT, nonfatal PE, all-cause mortality; per-protocol population) and the primary safety end point (major, postoperative bleeding events; safety population). The solid lines are the dose–response curves for rivaroxaban, estimated by logistic regression, including total daily dose as a covariate. The dotted lines represent the 95% confidence intervals for the primary efficacy end point, and the dashed lines represent the 95% confidence intervals for the primary safety end point. 20 B, Dose–response relationships between OD rivaroxaban and the primary efficacy end point (DVT, nonfatal PE, all-cause death; per protocol population) and the primary safety end point (major postoperative bleeding events; safety population). The solid lines are the dose–response curves for rivaroxaban, estimated by logistic regression, including total daily dose as a covariate. The dotted lines represent the 95% confidence intervals for safety. The hatched lines represent the 95% confidence intervals for efficacy. 22 BID indicates twice daily; DVT, deep vein thrombosis; OD, once daily; PE, pulmonary embolism.
In confirmatory phase III trials, rivaroxaban 10 mg OD demonstrated superior efficacy and similar safety to the SOC enoxaparin regimen after THR 23 and TKR. 24,25 Furthermore, rivaroxaban also demonstrated superior efficacy compared to the more intense enoxaparin regimen (30 mg BID) used in North America, with similar, low rates of major bleeding. 25 Thus, the selection of OD dosing for VTE prevention after THR/TKR surgery was based on solid preclinical and phase II evidence and confirmed by the results of the RECORD program.
Phase II Dose Ranging for VTE Treatment
Venous thromboembolism treatment regimens must satisfy 2 goals: near-term prevention of the extension (propagation) and embolization of the existing (index) clot after initial presentation and long-term prevention of the formation of new recurrent thrombi. As with VTE prevention after THR/TKR, the initial expectation was that BID dosing of rivaroxaban would prove optimal; therefore, a proof-of-principle study evaluated BID regimens (10-30 mg BID) versus enoxaparin followed by a vitamin K antagonist (VKA) in patients with symptomatic proximal DVT 26 to assess the ability of rivaroxaban to treat existing clots. On the basis of the knowledge derived from the orthopedic phase II program, a 40-mg OD dose was also included for rivaroxaban. Each treatment was administered for 12 weeks. The primary efficacy end point, improvement in clot resolution (reduction in thrombus burden) at 21 days was similar between all BID rivaroxaban doses and enoxaparin/VKA, although fewer patients receiving rivaroxaban 40 mg OD had improved resolution. Rates of major bleeding were low and similar across all treatment groups, except for a nonsignificant increase observed with rivaroxaban 30 mg BID. There were few recurrent venous thromboembolic events (a secondary end point) between 21 days and 12 weeks, with no results supporting one dosing regimen over another for the prevention of recurrent VTE. 26 Hence, these observations supported the start of VTE treatment without any prior LMWH dosing. The second phase II dose-ranging study compared the effectiveness of OD rivaroxaban regimens (20, 30, and 40 mg) for the prevention of recurrent VTE after 12 weeks of treatment with SOC LMWH followed by VKA. 27 The primary efficacy end point was the composite of symptomatic recurrent DVT, symptomatic PE, or asymptomatic deterioration in thrombotic burden at 12 weeks. Both the primary efficacy end point and the principal safety outcome (major plus clinically relevant nonmajor bleeding) occurred at lower rates than the LMWH/VKA comparator, with similar rates across the 3 rivaroxaban doses, suggesting that at the lowest rivaroxaban dose, 20 mg OD, efficacy had already plateaued. 27 Collectively, the results of these 2 phase II studies indicated improved resolution of the presenting clot with a BID rivaroxaban regimen, although no statistical evaluation was provided, and effective prevention of recurrent VTE occurred with both the OD and BID regimens. In both circumstances, the safety profile of rivaroxaban was similar to, or better than, that of the comparator regimen. 26,27 In addition to these results, earlier studies had suggested that more intense anticoagulation immediately after the index event might be beneficial. In the THRIVE 28 (Thrombin Inhibitor in Venous Thromboembolism) study evaluating ximelagatran against SOC in patients with acute DVT, the majority of the recurrent venous thromboembolic events occurred in the first few weeks of therapy (particularly in the ximelagatran arm; Supplementary Figure 1). Similarly, in the van Gogh studies comparing idraparinux with standard therapy in patients with DVT or PE, 29 most recurrent venous thromboembolic events occurred in the early weeks; idraparinux failed to meet noninferiority in the PE study, and the difference in the incidence of recurrence between the 2 groups originated mainly during the first 2 weeks of treatment (Supplementary Figure 2). Collectively, these findings indicated the need for more intense anticoagulation therapy in the early weeks. These considerations, together with the results of phase II studies, 26,27 provided good evidence in support of a 2-phase, single-drug treatment regimen to take forward into phase III trials of rivaroxaban for the treatment of VTE, that is, an initial BID phase followed by an OD maintenance regimen. 27 This proposed regimen was also consistent with population pharmacokinetic analyses (see Supplementary Material, section 3). 30
In summary, there was some evidence suggesting that a BID regimen would be optimal for reducing clot burden immediately after the index VTE but that for long-term prevention of recurrence, there was no apparent efficacy or safety advantage for either OD or BID dosing. Therefore, given the potential benefits of OD dosing, 10 –13 the lowest effective OD dose was chosen for the secondary prevention treatment phase. Hence, the specific regimen selected for phase III studies comprised an initial 3-week period of rivaroxaban 15 mg BID, followed by a maintenance dose for long-term prevention of VTE recurrence of 20 mg OD.
This regimen was successfully validated in phase III EINSTEIN program. In 2 phase III trials, EINSTEIN DVT 31 and EINSTEIN E, 32 rivaroxaban was noninferior to enoxaparin followed by VKA with regard to efficacy and had a similar safety profile. However, it is worth noting that in the EINSTEIN PE trial, the rate of major bleeding was significantly lower with rivaroxaban. 32 EINSTEIN DVT enrolled patients with DVT but without PE, whereas EINSTEIN PE enrolled patients with symptomatic PE, with or without DVT. The results confirmed the ability of rivaroxaban to provide effective VTE treatment in a single-drug approach, especially because, in both trials, fewer primary efficacy events occurred with rivaroxaban than with enoxaparin/VKA during the initial 3-week BID phase. 31,32 A third phase III trial, EINSTEIN EXT, 31 enrolled patients who had completed 6 to 12 months of treatment with either a VKA (from EINSTEIN DVT, EINSTEIN PE, or routine care) or rivaroxaban (from EINSTEIN DVT or EINSTEIN PE) and for whom the net benefit–risk of continuing with VKA therapy or with no further therapy was equivocal. Enrolled patients were randomized to rivaroxaban 20 mg OD or placebo for a treatment duration of 6 or 12 months. Compared with placebo, rivaroxaban significantly reduced the risk of recurrent VTE by 82%. Rates of major bleeding with rivaroxaban were low, with no significant difference between the treatment groups (4 patients in the rivaroxaban treatment group vs none in the placebo group), although the rates of clinically relevant nonmajor bleeding were increased. These results showed that rivaroxaban 20 mg OD could provide effective long-term prophylaxis against recurrent VTE in patients who had completed an initial course of therapy.
Thus, for this indication, careful phase II evaluation of different dose regimens, supported by pharmacokinetic modeling, identified a regimen optimized for both initial clot resolution followed by long-term prevention that, like the dosing for THR/TKR, was confirmed in a large phase III program. A new phase III study (EINSTEIN CHOICE; www.clinicaltrials.gov; NCT02064439) is currently underway, investigating a reduced dose (10 mg OD) and the standard dose (20 mg OD) of rivaroxaban for the long-term secondary prevention of recurrent VTE compared with acetylsalicylic acid (ASA) 100 mg OD.
Dose Selection for Stroke Risk Reduction in Patients With Nonvalvular AF
The mechanism of clot formation in the fibrillating atrium, particularly in the left atrial appendage, is believed to be similar to that for DVT. Both conditions are characterized by endothelial dysfunction, blood stasis, and a prothrombotic state and are associated with similar underlying clot pathophysiologies, which are predominantly red blood cell and fibrin rich. 33,34 Given that a dose-ranging study might expose patients receiving the lowest rivaroxaban dose to an unjustified increase in the risk of thromboembolic stroke, the decision was made to avoid such risk. This was possible owing to the availability of data from dose-finding studies for the treatment and prevention of VTE and the rationale that secondary prevention of VTE and prevention of stroke and systemic embolism in patients with nonvalvular AF are both chronic indications for which dose-adjusted VKA therapy is indicated. These similarities meant that no phase II study was conducted for stroke risk reduction in patients with AF.
The key aim of antithrombotic therapy in AF is to prevent the formation of clots within the atria (usually in the left atrial appendage) that can subsequently embolize and result in cardiogenic ischemic stroke. The dose selection for use in patients with nonvalvular AF was rivaroxaban 20 mg OD, based on the results of phase II VTE treatment studies. 26,27 A total daily dose of 20 mg was the lowest evaluated in these studies, but both cases yielded efficacy and safety results similar to those seen with higher doses. An OD regimen was selected based on the observation that OD and BID dosing had similar efficacy and safety profiles with respect to the prevention of recurrent VTE, but with OD dosing seeming to have a more favorable bleeding profile than BID dosing compared with the comparator regimen. 26,27 Therefore, this dose regimen was also considered to offer the best balance of efficacy and safety considering the advanced age and comorbidities common among patients with AF. 35
In addition, the above-mentioned dose selection for stroke prevention in patients with AF was also supported by pharmacokinetic modeling of simulated patients with AF, 30 after taking into consideration that the participants enrolled in phase II VTE treatment studies differed demographically from most patients with AF. To assess the pharmacodynamics of rivaroxaban in a population of patients with AF, a simulated cohort was used to reflect its demographics, including the greater age and reduced renal function common in these patients. The population pharmacokinetic analyses from phase II VTE treatment trials, as discussed previously, showed that age and renal function had moderate effects on rivaroxaban clearance. 30 The cohort was constructed by modifying the demographics of the VTE treatment population to reflect those of an AF population, based on the demographics of patients with AF enrolled in the earlier SPORTIF (Stroke Prevention using ORal Thrombin Inhibitor in atrial Fibrillation) III and SPORTIF V phase III trials. 30 The simulation showed that moderate renal impairment (CrCl 30–49 mL/min) led to a modest increase in rivaroxaban exposure, with age having a smaller effect, although the Cockcroft-Gault formula includes corrections for age, weight, and gender. 36 The modest increase in exposure was expected in light of the earlier demonstration of reduced clearance of rivaroxaban associated with renal impairment. 9 However, the simulation suggested that it could be corrected by reducing the rivaroxaban dose from 20 mg OD to 15 mg OD (Figure 5). 30 Hence, the rivaroxaban dosing regimen taken forward to phase III ROCKET AF (Rivaroxaban Once daily, Oral, Direct Factor Xa Inhibition Compared with Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation) was 20 mg OD in patients with normal or mildly impaired renal function (CrCl ≥ 50 mL/min) and 15 mg OD in patients with moderate renal impairment (CrCl 30–49 mL/min). 35 Thus, this dosing regimen was based on evidence drawn from both empirical phase II studies and extensive pharmacokinetic modeling studies.
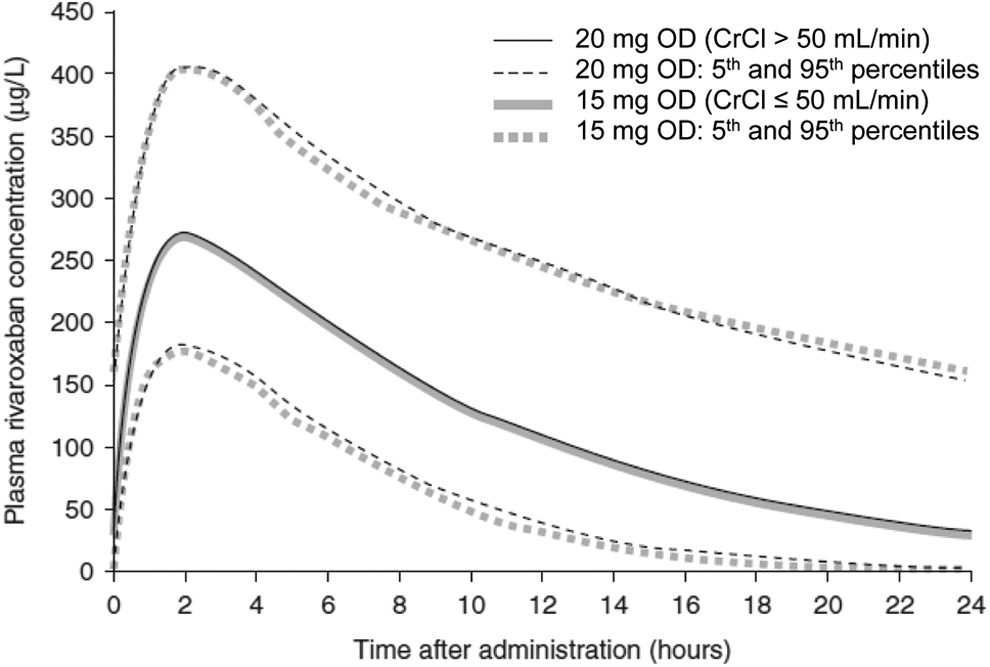
Plasma rivaroxaban concentration–time profiles for the simulated virtual atrial fibrillation population. 30 Results are shown for patients with normal renal function (CrCl > 50 mL/min) receiving rivaroxaban 20 mg once daily and for patients with renal impairment (CrCl ≤ 50 mL/min) receiving rivaroxaban 15 mg OD. CrCl indicates creatinine clearance; OD, once daily.
The ROCKET AF phase III study validated the selected dosing regimens. In the intention-to-treat analysis, rivaroxaban demonstrated noninferior efficacy versus warfarin with respect to the primary end point (composite of stroke or systemic embolism) and superiority in the prespecified on-treatment analysis. Results for both major or clinically relevant nonmajor bleeding (the principal safety outcome) and major bleeding were similar between the 2 treatment groups. 37 Event rates were higher in patients with moderate renal impairment, as expected, but there was no evidence of any difference in treatment effect (rivaroxaban vs warfarin) compared to patients having normal renal function (Supplementary Table 3 and Supplementary Material, section 4). 38
Dose Selection for the Secondary Prevention of Recurrent Atherothrombotic Events After an ACS
Clinical trials with the antiplatelet agents prasugrel and ticagrelor reported a residual risk of approximately 10% of experiencing a major cardiovascular event (death from cardiovascular causes, myocardial infarction, or stroke) for the subsequent 12 to 15 months after an ACS event. 39,40 The dual pathway of thrombus formation in ACS, involving platelets and fibrin, suggested a potential role for a factor Xa inhibitor to reduce this risk. 41
The ATLAS ACS TIMI 46 (Anti-Xa Therapy to Lower cardiovascular events in Addition to aspirin with/without thienopyridine therapy in Subjects with Acute Coronary Syndrome) phase II dose-escalation study investigated rivaroxaban 5, 10, and 20 mg doses given OD or the same total daily dose given BID in combination with ASA or ASA plus a thienopyridine (clopidogrel or ticlopidine). 42 Rivaroxaban was associated with a dose-dependent increase in bleeding, but with the efficacy already noted at lower doses, and the lowest 2 doses (5 and 10 mg total daily dose) were consequently selected for further investigation in the ATLAS ACS 2 TIMI 51 (Anti-Xa Therapy to Lower cardiovascular events in Addition to aspirin with/without thienopyridine therapy in Subjects with Acute Coronary Syndrome) phase III trial. 43 Twice-daily dosing was selected over OD dosing on the basis that the pharmacokinetic and pharmacodynamic profiles suggested insufficient trough levels for OD doses below 10 mg total daily doses. Furthermore, 2.5 mg BID and 5 mg BID seemed to offer the best balance between safety and efficacy. 42
In the ATLAS ACS 2 TIMI 51 study, rivaroxaban in combination with ASA alone or ASA plus clopidogrel or ticlopidine significantly reduced the risk of the composite primary efficacy outcome of death from cardiovascular causes, myocardial infarction, or stroke (P < .05) but increased the risk of Thrombolysis In Myocardial Infarction major bleeding (not related to coronary artery bypass graft surgery) and intracranial hemorrhage (although not fatal bleeding), compared with placebo. 43 Rivaroxaban 2.5 mg BID was associated with a more favorable overall benefit–risk profile (with lower cardiovascular and all-cause mortality as well as lower bleeding rates) compared with rivaroxaban 5 mg BID. 44 This result led to the European approval of the 2.5 mg BID dosing regimen for this indication in combination with ASA alone or ASA plus clopidogrel or ticlopidine for patients after an ACS event who have elevated cardiac biomarkers and no history of prior stroke/transient ischemic attack. 1
Once Daily Versus Twice Daily: Safety Implications
Although it is commonly believed that anticoagulant-associated bleeding is associated with Cmax, empirical data do not uniformly confirm this belief. In the case of rivaroxaban, the close correlation between pharmacokinetic and pharmacodynamics suggested that bleeding was likely to be associated with Cmax. However, in 2010, Weitz et al published a phase II study of edoxaban for stroke risk reduction in patients with AF. 45 Edoxaban (formerly DU-176b) is another oral direct factor Xa inhibitor approved for the treatment and secondary prevention of VTE and the prevention of stroke in patients with AF having one or more risk factors 46 and has similar pharmacokinetics to rivaroxaban; specifically, time to Cmax for edoxaban is 1 to 2 hours and elimination t1/2 is approximately 8 to 10 hours. 45 The phase II edoxaban study evaluated 4 dosing regimens: 30 and 60 mg OD and 30 and 60 mg BID. It was observed that for the same total daily dose of 60 mg, all bleeding events (all bleeding, major plus nonmajor clinically relevant bleeding, and major bleeding) occurred at a lower rate with the 60-mg OD regimen than with the 30-mg BID regimen (Table 2). 45
Bleeding Events in a Phase II Study of Edoxaban in Patients With Atrial Fibrillation. 45,a
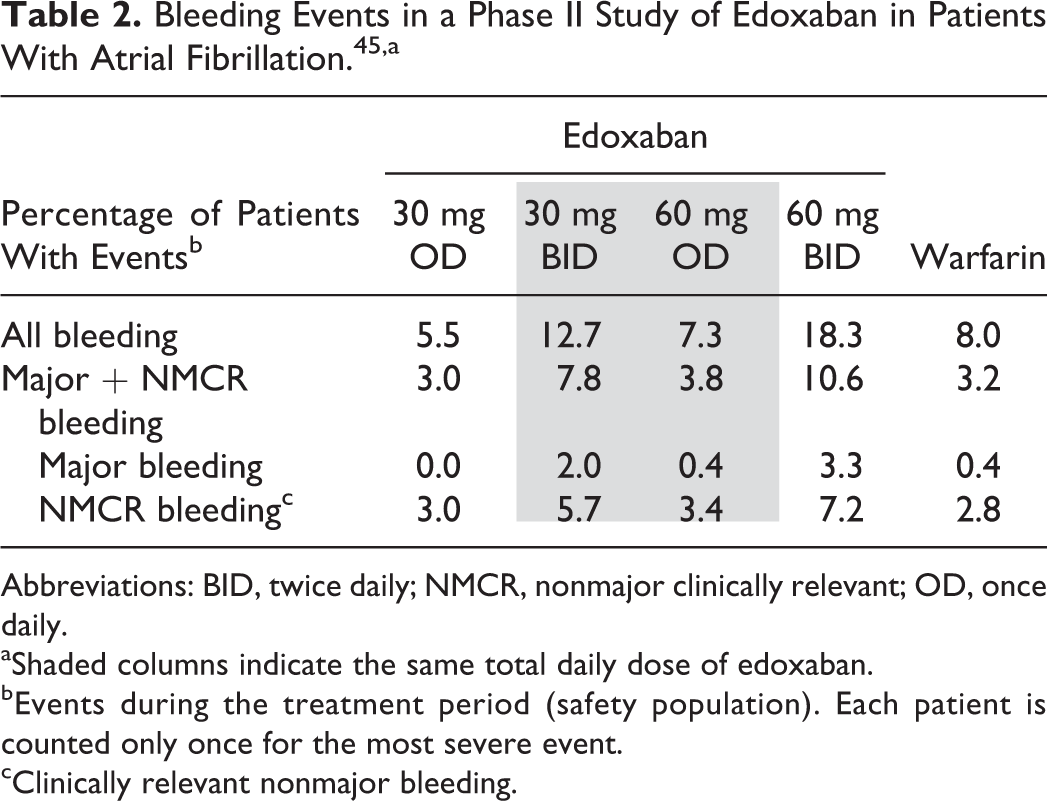
Abbreviations: BID, twice daily; NMCR, nonmajor clinically relevant; OD, once daily.
aShaded columns indicate the same total daily dose of edoxaban.
bEvents during the treatment period (safety population). Each patient is counted only once for the most severe event.
cClinically relevant nonmajor bleeding.
Pharmacokinetic analyses of edoxaban exposure in this study showed that, as expected, Cmax was greater with 60 mg OD than with 30 mg BID and AUC was the same for both regimens. However, minimum plasma drug concentration (Cmin) was notably lower with 60 mg OD than with 30 mg BID, suggesting that bleeding rates correlated more with trough concentrations and not with peak plasma levels. 45 This finding was supported by a subsequent simulation of edoxaban exposure–response relationships in patients with AF. 47 This study demonstrated a strong correlation between bleeding rates and Cmin levels (Figure 6) and concluded that steady-state trough levels (Ctrough) were the best predictor of bleeding events. 47
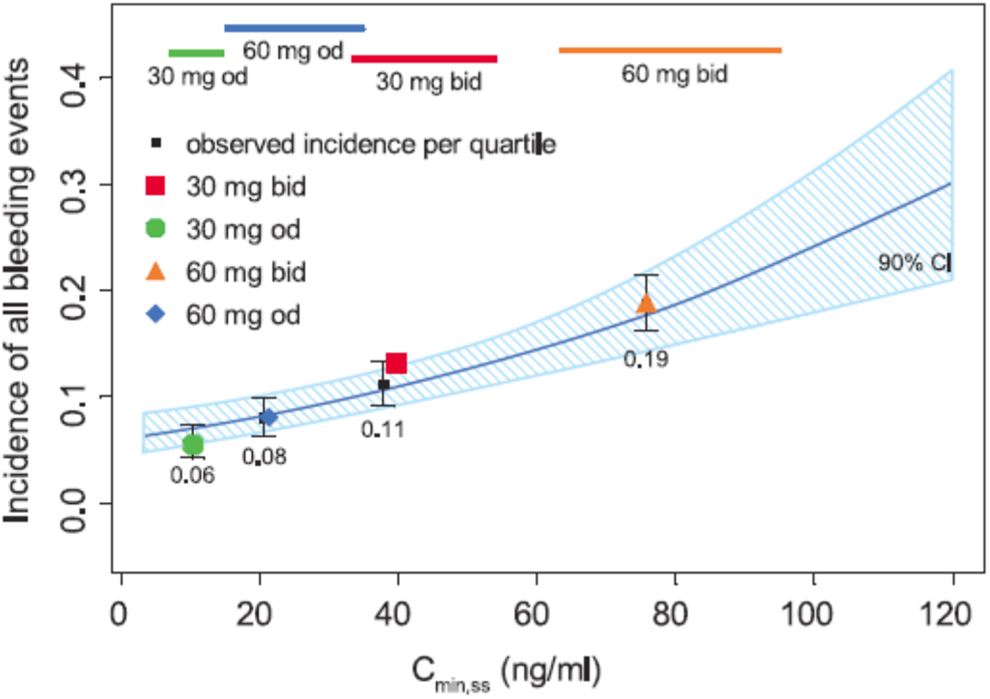
Goodness-of-fit of logistic regression model for rates of bleeding versus Cmin for edoxaban. 47 The solid blue line represents the logistic model prediction, and the blue shaded area represents the 90% CI. Horizontal colored bars represent the 10th to 90th percentiles of Cmin,ss by treatment group. The colored symbols represent the incidence of bleeding events by edoxaban treatment group and are plotted at the median Cmin,ss for each treatment group. Black squares and whiskers represent the computed median and ±1 standard error of incidence of bleeding events for each quartile of Cmin,ss across all 4 dosages. BID indicates twice daily; CI, confidence interval; Cmin, minimum plasma drug concentration; Cmin,ss steady state minimum plasma drug concentration; OD, once daily.
Collectively, these results suggest that bleeding rates may depend on the time for which plasma levels of an anticoagulant exceed a certain threshold, which is likely to be longer in duration for a BID regimen than for the same total daily dose delivered OD. In either event, these studies indicate that the risk of bleeding is likely to be lower with OD than with BID regimens, by virtue of the lower Ctrough. 45
Based on these findings, the 2 edoxaban OD dose regimens (30 and 60 mg) were taken forward and were validated in a phase III study (ENGAGE-AF [Effective Anticoagulation with Factor Xa Next Generation in Atrial Fibrillation–Thrombolysis in Myocardial Infarction 48]), in which they were noninferior to warfarin in the intention-to-treat analysis with regard to the primary efficacy end point of stroke and systemic embolism and were associated with significantly lower rates of major bleeding. 48
Although evidence supporting OD dosing of rivaroxaban in appropriate indications has also been validated in phase III studies, it is less clear why other factor Xa inhibitors, for example apixaban, are dosed BID in the same indications. Unfortunately, there are limited data available on this issue at present, and where OD/BID comparisons have been published for apixaban, event rates have been too low to draw any definitive conclusions. 49,50 However, all the licensed novel oral anticoagulants have demonstrated a positive benefit–risk profile in clinical studies, for example, for stroke prevention in patients with nonvalvular AF (given OD or BID), although a direct comparison of their efficacy and safety is not appropriate owing to the different study designs, patient populations, and outcome definitions used. Information in this area is steadily being acquired, for example, OD dosing regimens have been associated with improved adherence compared with drugs taken BID in routine clinical practice. 51,52 Alongside adherence, persistence to therapy is also likely to be better with OD versus BID regimens. Supporting evidence has been seen in several real-world studies: Persistence and discontinuation among patients with nonvalvular AF on rivaroxaban was improved relative to dabigatran in the United States 53 ; an analysis of health care claims data reported a significantly higher proportion of patient adherence to rivaroxaban therapy (72.7%) compared with dabigatran (67.2%; P < .001) or apixaban (69.5%; P < .001) 54 ; and discontinuation with rivaroxaban was 13.6 per 100 patient-years versus 25.8 per 100 patient-years with dabigatran in the Dresden NOAC registry. 55,56 Taken together, OD dosing may be one of the contributing factors to the higher adherence and persistence seen with rivaroxaban compared with dabigatran or apixaban, which are given as BID regimens.
Discussion
The guiding principle throughout the development of the different rivaroxaban dosing regimens (Table 1) has been regimen selection based on careful and comprehensive empirical phase II studies, supported by rigorous pharmacology studies.
In accordance with a priori expectations, rivaroxaban clinical development was initiated with BID regimens. Data indicating that OD use might be feasible developed gradually, and each step in the development of a dosing regimen for clinical use was evidence based, that is, well supported by experimental data. Thus, selection of each dosing regimen was data driven, and each regimen was developed for a specific indication, based on phase II studies supported by pharmacology studies as appropriate. Furthermore, with regard to the latter, a number of parameters in addition to elimination t1/2 were evaluated to support the final dosing decision.
Optimization of dosing regimens requires consideration of both efficacy and safety outcomes. In the majority of indications for which rivaroxaban is the sole intervention, empirical data have shown OD regimens to offer the best balance between efficacy and safety, with good results for each when rivaroxaban was compared to the respective comparators in phase III studies. Furthermore, in ENGAGE-AF, the OD dosing of edoxaban demonstrated noninferiority to warfarin in the prevention of stroke in patients with AF. 48
The exception has been the demonstration that a BID regimen is optimal for the initial treatment of acute VTE, supported by good evidence that a more intensive regimen is optimal during this initial treatment phase. The reasons are not immediately clear, but because Ctrough for BID administration can be expected to be at least as great as Ctrough for OD administration, 2 BID administration is likely to maintain a higher median concentration than OD administration. Because the main objective for this initial phase is resolution of the existing clot and prevention of further propagation, it may be that a higher median concentration of rivaroxaban (ie, mass action effect) is required to facilitate the diffusion of rivaroxaban throughout the fibrin mesh of the clot. Additionally, more frequent administration can be expected to achieve steady state quicker following the index event.
Twice-daily administration is also optimal when a much lower dose of rivaroxaban, that is, 2.5 mg BID, is administered with concomitant antiplatelet therapy for the prevention of recurrent atherothrombotic events after ACS, 42,43 suggesting that the optimal dosing regimen may depend not only on the particular disease pathology but also on the overall combination of antithrombotic drugs used for treatment. Furthermore, data cited previously (Figure 1) 7,8 indicate that when the total daily dose of rivaroxaban is 10 mg or more, OD dosing may be feasible. However, when the total daily dose is less, BID dosing will likely be required because OD doses of 5 mg or lower would be unlikely to provide sufficient trough coverage over 24 hours, that is, to maintain an adequate pharmacodynamic effect. This is consistent with the observation that ETP is reduced 24 hours after 30 mg of rivaroxaban but not after 5 mg (Figure 1), 7,8 thus providing a further, indirect support for a BID regimen when such a low dose is to be used, as is the case after an ACS event.
It was not immediately apparent why OD rivaroxaban regimens would offer the optimal balance of efficacy and safety in light of the relatively short t1/2 and the conventional wisdom that bleeding was most likely when pharmacodynamic effects were maximal, ie, at Cmax. However, the demonstration from the edoxaban phase II studies 45,47 that bleeding rates correlated with Ctrough levels (Figure 6), and not with Cmax, provides a cogent explanation as to why OD regimens might offer a better balance between efficacy and safety than BID regimens, although the particular attributes of individual drugs should always be considered.
Although a single anticoagulant, such as rivaroxaban, can provide clinical benefit in multiple therapeutic indications, this does not mean that the same dosing regimen will provide the optimal balance of benefit and risk in all indications. Dosing regimens should be optimized for each indication. This article provides an historical overview of the development of the different regimens approved for rivaroxaban in different indications, each of which has been thoroughly validated in a phase III registration trial. This summary should facilitate an understanding as to why different dosing regimens for the same drug are appropriate in different therapeutic indications.
Footnotes
Acknowledgments
The authors would like to acknowledge Yong-Ling Liu, Chameleon Communications International, who provided editorial assistance with funding from Bayer HealthCare Pharmaceuticals and Janssen Scientific Affairs, LLC.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Dr Scott D. Berkowitz is an employee of Bayer HealthCare Pharmaceuticals, Inc. Dr Dagmar Kubitza is an employee of Bayer Pharma AG. Dr Frank Misselwitz is an employee of Bayer Pharma AG with significant stock ownership in Bayer shares.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Bayer Pharma AG.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.