
Abstract
Acute coronary syndrome (ACS) encompasses a spectrum of diseases, ranging from ST-elevation myocardial infarction to non-ST-elevation myocardial infarction and unstable angina. A key initiating event in the pathology of ACS is atheromatous plaque disruption, in which the exposure of thrombogenic material triggers simultaneous activation of primary and secondary hemostatic pathways. Targeting platelet-mediated thrombus formation with dual antiplatelet therapy comprising acetylsalicylic acid and a P2Y12 antagonist is the current mainstay for management of ACS. However, a significant proportion of patients remain at risk of cardiovascular events. Fibrin is an important contributor to thrombogenesis and may account for the residual event rates. This review examines evidence for the role of the coagulation cascade in thrombus formation in ACS, which provides a rationale for the use of anticoagulation therapy. The current status of research with novel oral anticoagulants in combination with dual antiplatelet therapy for the secondary prevention of ACS is also discussed.
Introduction
The clinical burden of acute coronary syndrome (ACS) is substantial. The American Heart Association reports that each year approximately 785 000 Americans will experience their first coronary attack and approximately 470 000 Americans will have a recurrent episode. 1 In 2007, there were 1 172 000 unique hospitalizations for ACS reported in the United States, of which 731 000 were for myocardial infarction (MI) alone and 431 000 were for unstable angina (UA) alone. 1 As higher sensitivity troponin tests are introduced, enabling the diagnosis of MI at lower cutoff points, 2,3 the incidence of non-ST-elevation myocardial infarction (NSTEMI) is expected to rise and the incidence of UA to fall.
Atherothrombosis (Figure 1), defined as atherosclerotic plaque disruption with superimposed 4 thrombus, is the pathophysiologic hallmark of ACS. Platelets play a crucial role in the development of thrombotic complications, and targeting platelet aggregation with antiplatelet agents has proved successful in ACS. 5 Monotherapy with acetylsalicylic acid (ASA), a platelet cyclooxygenase inhibitor, has been superseded by dual antiplatelet therapy that combines ASA with clopidogrel (a thienopyridine P2Y12 antagonist that inhibits the adenosine diphosphate [ADP]-dependent pathway of platelet activation), and more recently prasugrel 6 or ticagrelor 7 (Figure 2). The CURE study 12 demonstrated that combining clopidogrel 8 –11 and ASA further reduced the incidence of recurrent events in patients with NSTEMI ACS at 1 year compared with ASA alone (9.3% vs 11.4% in the clopidogrel plus ASA group and placebo plus ASA group, respectively [relative risk of 0.80; 95% confidence interval (CI) 0.72-0.90; P < .001]). Extended use (up to 1 year) of dual antiplatelet therapy of ASA with a P2Y12 antagonist has become the “gold standard” for the prevention of recurrent ischemia after an ACS event. 13 –15 Prasugrel is a thienopyridine that inhibits ADP-induced platelet aggregation more robustly than clopidogrel. 16 Data from the TRITON-TIMI 38 study 6 showed that cardiac events over 15 months occurred in 12.1% of the patients receiving clopidogrel and 9.9% of the patients receiving prasugrel (hazard ratio [HR] for prasugrel vs clopidogrel 0.81; 95% CI 0.73-0.90; P < .001). Ticagrelor is a reversible P2Y12 receptor antagonist in a chemical class distinct from the thienopyridines. 17 In the PLATO trial, 7 ischemic events (including fatal events) were reported in 9.8% of the patients receiving ticagrelor, compared with 11.7% of those receiving clopidogrel at 1 year (HR = 0.84; 95% CI 0.77-0.92; P < .001). Taken together, these dual antiplatelet trials report a residual risk of recurrent ischemic events of approximately 10%, suggesting that, despite moderate improvements with newer agents like prasugrel and ticagrelor, treatment with antiplatelet agents may have reached a therapeutic “plateau.”
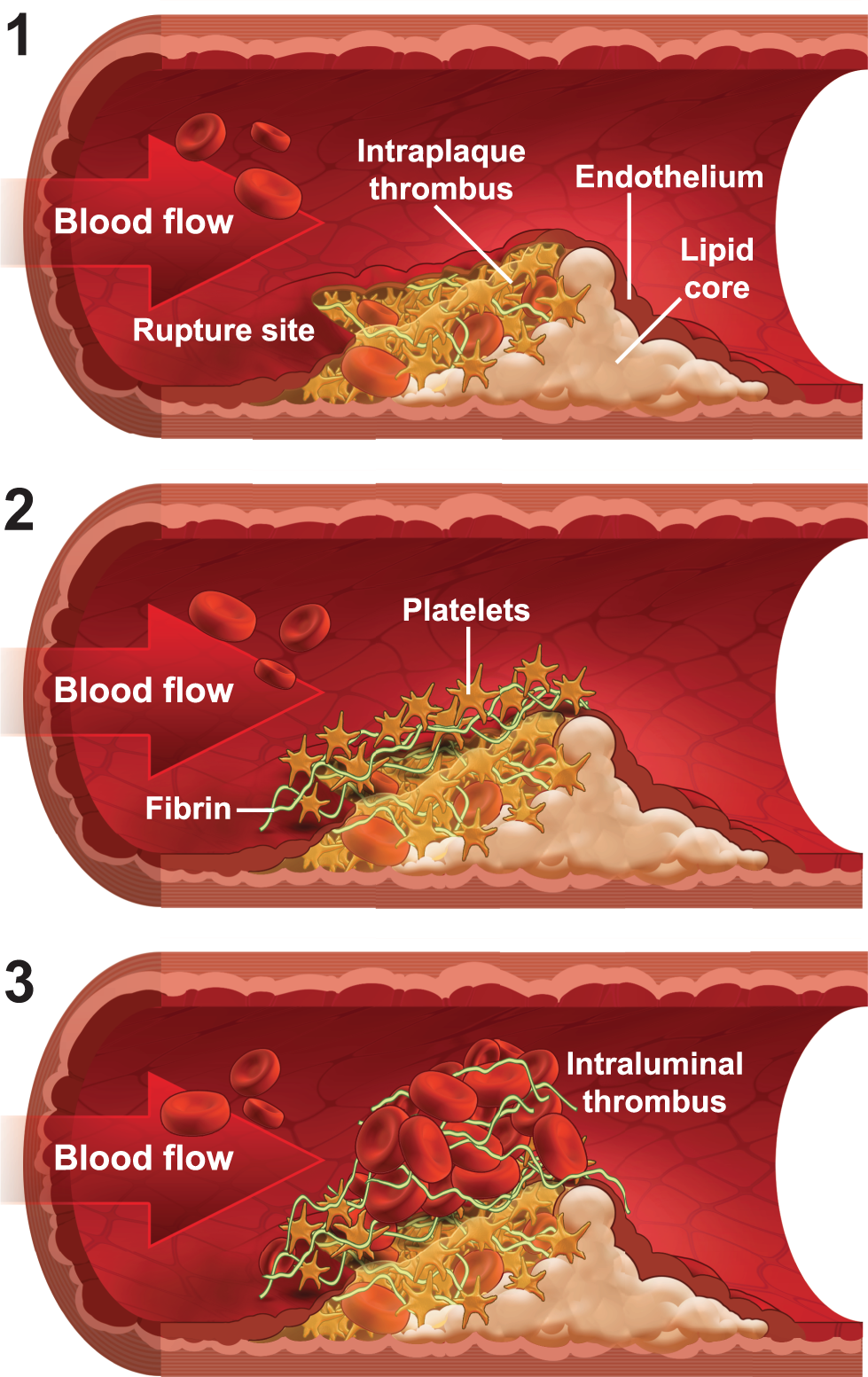
Schematic representation of thrombus development after plaque disruption. Initially, platelets are more abundant than fibrin within the plaque itself; at the rupture site, a layer of platelets covers a region of dense fibrin. Later, fibrin and erythrocytes become enmeshed and trapped in the thrombus, leading to thrombus enlargement and vessel occlusion. 4
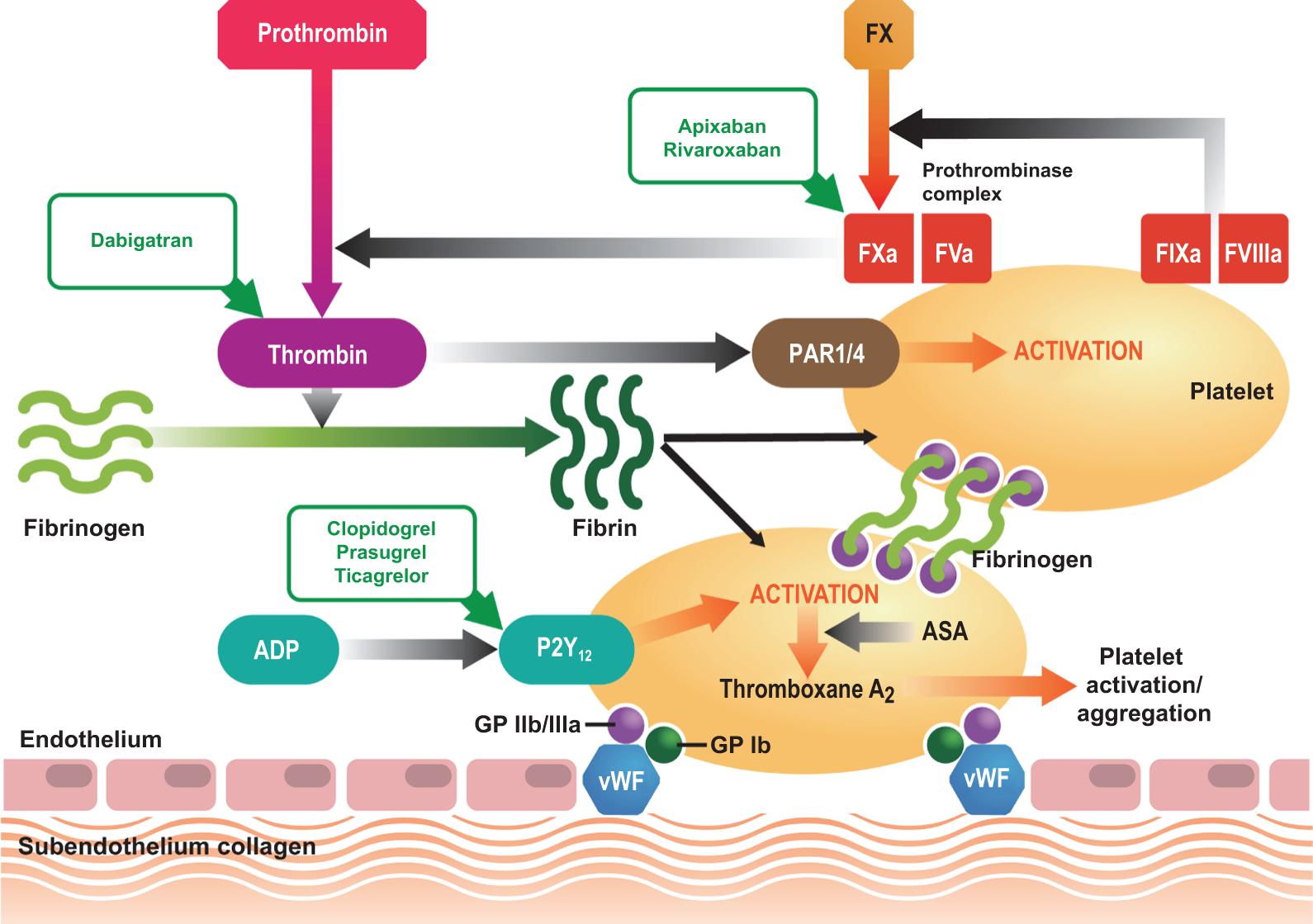
Simplified view of the primary and secondary hemostatic responses to vascular injury and the commonly used or trialed antithrombotic agents in patients with acute coronary syndrome (ACS). Endothelial damage exposes subendothelial structures that trigger interactions between glycoprotein (GP) receptors on platelets and various adhesion proteins such as von Willebrand factor (vWF) and collagen. 8 These interactions activate platelets at the site of damage, which release granules that in turn lead to further platelet activation and aggregation. 9,10 Fibrinogen binds to GP IIb/IIIa receptors on adjacent platelets, partially stabilizing the thrombus. After endothelial damage, tissue factor (TF)-bearing cells (eg, those in the blood vessel wall) generate a small amount of thrombin via the factor (F) VIIa–TF complex (not shown); this small amount of thrombin triggers the formation of the prothrombinase complex (FXa–Va) on the surface of activated platelets, via the tenase (FIXa–VIIIa) complex. 10,11 This, in turn, generates a “burst” of thrombin that catalyzes the conversion of fibrinogen to fibrin. In humans, the thrombin generated also binds to protease-activated receptors (PARs) 1 and 4, leading to further platelet activation. For simplicity, the additional functions of thrombin (eg, activation of factors XI and XIII, and thrombomodulin) are not shown. The sites of action for the antiplatelet agents acetylsalicylic acid (ASA), clopidogrel, prasugrel, and ticagrelor; the direct thrombin inhibitor dabigatran; and the direct factor Xa inhibitors apixaban and rivaroxaban are shown.
Under normal physiologic conditions, blood vessel integrity after injury is maintained by primary and secondary hemostatic mechanisms via the aggregation of platelets and deposition of fibrin. Although the clinical utility of targeting the former mechanism with antiplatelet therapy is established, anticoagulants are not usually given to patients long term (unless they are for a comorbid condition such as atrial fibrillation [AF]), 13,14 and the role of fibrin in atherothrombosis is generally overlooked.
In this review, both the molecular and clinical rationale are presented for targeting the fibrin as well as the platelet component of clot formation and, by doing so, potentially improving the current levels of secondary prevention.
Pathophysiology of ACS
The fully developed human atherosclerotic plaque is designated by the American Heart Association as type IV (atheroma) or type Va (fibroatheroma). 18 Type IV lesions may not cause much occlusion of the arterial lumen but they have great clinical significance because their surface is prone to tearing or erosion. 18 The size of the plaque does not appear to be as important a predictor of the risk of plaque rupture as the composition or “vulnerability” of the plaque itself. 19 Intraplaque hemorrhage from the vasa vasorum supplying blood to the plaque has been implicated as a critical event in the induction of plaque instability. 20 Other systemic factors that may contribute to plaque rupture are increased coagulability and systemic inflammation. 21 Rupture of the fibrous cap occurs in approximately three-quarters of ACS culprit lesions; 22 other mechanisms that account for the remaining events include superficial erosion. In patients with sudden cardiac death, pathologic studies revealed that ACS culprit lesions were ruptured plaque in 75% and eroded plaque in 25% of the cases. 23
The lipid core of a ruptured plaque is highly thrombogenic. 24 Initially, an intraplaque thrombus rich in platelets is formed, with fibrin and red blood cells present in lesser amounts (Figure 1). 4 The second stage involves a transitional zone at the site of rupture where there is an area of densely packed fibrin covered by a layer of platelets. The thrombus itself may become exposed to the blood flow in the artery (mural thrombus) and, in the final stage, may extend into the lumen. This intraluminal thrombus is composed predominantly of fibrin and platelets, whereas distal to the thrombus is a loose fibrin network with enmeshed erythrocytes and few platelets.
Transient or partial occlusion associated with UA and NSTEMI is usually the result of mural thrombus formation composed predominantly of platelets and some fibrin. 4,25 In contrast, ST-elevation myocardial infarction (STEMI) usually occurs when a fibrin-rich thrombus completely occludes a coronary artery, leading to often extensive myocardial necrosis. 25,26 However, evidence suggests that, even in the “milder” presentations of ACS (eg, UA), fibrin can form a major component of clots. Angioscopic evaluation of coronary artery thrombi in patients with ACS revealed that almost a third of patients with UA had fibrin-rich thrombi. 27
Platelet and Fibrin Activation in Thrombus Formation: Underlying Mechanisms
Primary hemostasis (triggered by plaque rupture under pathophysiologic conditions) is characterized by platelet aggregation and formation of an unstable plug. Secondary hemostasis involves a complex interaction between platelet membranes and coagulation factors that ultimately stabilizes the platelet plug (Figure 2). Although the classic view of thrombus formation paints a picture of disparate hemostatic processes converging to form a stable plug, real-time imaging has shown that platelet and fibrin deposition occurs nearly simultaneously following experimentally induced endothelial injury. 28
In addition to plaque rupture, an imbalance in secondary hemostatic mechanisms, marked by increased blood thrombogenicity, may also play a key role in the onset of ACS. 29 Several hemostatic factors have been implicated in the increased thrombogenicity of blood in patients with ACS. For example, tissue factor is active within the thrombus and is associated with intrathrombus fibrin generation. 30 Additionally, plasma concentrations of tissue factor and expression levels of tissue factor in platelets are significantly increased in patients with ACS. 31,32 A “hypercoagulable state” is also known to persist long after clinical stabilization. 33
At the crossroads of the primary and secondary hemostatic pathways, thrombin acts as a central player in thrombus formation through its potent activation of platelets 34 and by catalyzing the conversion of fibrinogen to fibrin. Thrombin enhances platelet activation by cleaving the protease-activated receptors (PARs) 1 and 4 on the platelet surface (Figure 2); however, the binding of thrombin to a glycoprotein Ib-dependent signaling pathway also activates platelets through a fibrin-dependent mechanism. 35,36 This signaling pathway might, therefore, account for the residual risk of thrombosis associated with the use of antiplatelet regimens.
Combined Anticoagulant and Antiplatelet Therapy may Improve Outcomes in ACS
To improve the clinical outcomes, therapeutic management strategies in ACS have been looking beyond targeting solely the platelet-driven pathway. The observation that the coagulation pathway in patients with ACS shows an increase in basal level activity for at least 6 months after clinical stabilization 33 has important implications for the treatment of patients with ACS. Furthermore, the inability of antiplatelet agents to inhibit the fibrin-mediated pathway may account for the failure of these agents to achieve maximal efficacy in reducing ischemic events in ACS, and provides a clear mechanistic rationale for assessing the use of anticoagulants in ACS.
Coagulation factors VII, IX, and X are vitamin K-dependent enzymes whose synthesis is blocked by the actions of vitamin K antagonists (VKAs) such as warfarin. 37 Studies of VKAs in patients with ACS have shown a substantial incremental benefit of long-term VKA therapy when added to ASA. 38,39 A meta-analysis of trials in patients with ACS demonstrated a reduction in mortality of 18%, MI of 52%, and stroke of 53% with moderate-intensity VKA therapy (international normalized ratio of 2.0-3.0) compared with control therapy. 40 However, these benefits were associated with an almost 8-fold increased risk of major bleeding among those treated with VKAs compared with controls. 40 Furthermore, a number of studies have shown that the use of dual antiplatelet therapy in stenting was associated with fewer bleeding events and a more favorable efficacy profile than VKA therapy or VKA plus ASA therapy. 41 –45 There are also practical limitations associated with VKAs to consider, which include considerable interpatient variability in dose response, numerous food and drug interactions, a slow onset and offset of action, a narrow therapeutic index, and an increased risk of bleeding. 37
Novel oral anticoagulants that selectively target single molecular components of the coagulation cascade have been trialed as potential therapeutic alternatives to VKAs. Dabigatran, apixaban, rivaroxaban, and darexaban represent 4 such novel oral anticoagulants that are either approved or are currently or have previously been in development for a number of thromboembolic indications, including the secondary prevention of ischemic events in ACS. These 4 oral anticoagulants have been evaluated in combination with antiplatelet therapy for the prevention of recurrent ischemic events in ACS in clinical trials.
Dabigatran, a direct thrombin inhibitor, specifically and reversibly inhibits both free and clot-bound thrombin by binding to the active site of the thrombin molecule.
46
The mechanism of action of dabigatran prevents the feedback amplification of the coagulation pathway, in effect blocking the molecular cascade that leads to fibrin generation; furthermore, thrombin-mediated platelet activation is also affected (Figure 2). Dabigatran has recently been approved for stroke prevention in patients with AF in the United States,
47
Canada,
48
and the European Union.
49
This follows the results of the phase III RE-LY clinical trial, which compared 2 doses of dabigatran (110 and 150 mg twice daily [bid]) with dose-adjusted warfarin in patients with AF and at least 1 risk factor for stroke. In the phase II RE-DEEM study,
50
1861 patients with recent MI were randomized to 1 of 4 doses of dabigatran or placebo. Patients were already receiving the standard dual antiplatelet therapy of ASA and clopidogrel. Although the study was not powered for robust statistical comparison of clinical outcomes, there was a dose-dependent increase in the primary outcome event rate (composite of major or clinically relevant minor bleeding) with dabigatran compared with placebo, although the absolute incidence was low (3.5%-7.9% for dabigatran 50-150 mg bid vs 2.2% for placebo). Concentrations of 1 marker of thrombotic burden,
The development of other agents has focused on the inhibition of factor Xa. Because of its position in the coagulation cascade and its pivotal role in large-scale thrombin generation, the inhibition of factor Xa would be expected to substantially reduce the activation of the coagulation cascade and of thrombin-mediated platelet activation. However, it has been suggested that marked factor Xa suppression can occur without adversely affecting hemostasis, because factor Xa plasma activity must be 10% of normal levels or lower to register even as a mild deficiency. 54
Apixaban is a selective and direct inhibitor of both free and clot-bound factor Xa (Figure 2). 55 In the phase II dose-ranging APPRAISE study in patients with ACS (n = 1715), 56 apixaban at doses of 2.5 mg bid, 10 mg once daily (od), 10 mg bid, or 20 mg od was added to antiplatelet therapy (ASA or ASA and clopidogrel). A dose-dependent increase in bleeding was seen in patients receiving apixaban, together with a nonsignificant trend toward a reduction in ischemic events. Excess total bleeding was observed in the 10 mg bid and 20 mg od arms, and these arms were discontinued on the advice of the study’s Data Monitoring Committee. Notably, the increase in bleeding was more pronounced, and the reduction in ischemic events was less evident, with apixaban in those receiving ASA plus clopidogrel (76% of patients) than in those taking ASA alone; it was hypothesized that the safety and efficacy of apixaban might vary depending on the background antiplatelet therapy that patients received. However, the larger phase III APPRAISE-2 study, which evaluated apixaban 5 mg bid versus placebo in patients post ACS (the majority of whom also received clopidogrel and ASA), was terminated early after 7392 patients had been enrolled. 57 This was because of an associated increase in major bleeding events (including intracranial and fatal bleeding events) that was not offset by clinically meaningful reductions in recurrent ischemic events. There was no significant reduction in the annual rate of cardiovascular death, MI, or ischemic stroke with apixaban (13.2 events per 100 patient-years) versus placebo (14.0 events per 100 patient-years; HR = 0.95; 95% CI 0.80-1.11). Major bleeding was more common with apixaban; intracranial hemorrhage rates were also significantly higher (0.6 vs 0.2 events per 100 patient-years; HR = 4.06; 95% CI 1.15-14.38; P = .03). There were 5 fatal bleeds in the apixaban arm compared with none in the placebo arm. Although these findings were clearly disappointing, the underlying reason may have been that the dose of apixaban selected was too high for this indication; further study may, therefore, be merited.
Rivaroxaban is another oral, direct factor Xa inhibitor that also inhibits both free and clot-bound factor Xa and impedes thrombin generation via inhibition of the prothrombinase complex (Figure 2). 58,59 The phase II ATLAS ACS TIMI 46 trial was a large dose-ranging study that evaluated rivaroxaban for the prevention of secondary ischemic events in patients with ACS (n = 3491). 60 In this study, the addition of rivaroxaban to either single (n = 761) or dual (n = 2730) antiplatelet therapy over a 6-month study period resulted in further reductions in ischemic events and an increase in bleeding compared with single or dual antiplatelet therapy alone. Based on the efficacy and safety profile, the lowest doses tested, 2.5 and 5 mg bid, were subsequently carried forward to the phase III ATLAS ACS 2 TIMI 51 trial. 60,61 A broad population of patients with ACS were enrolled into this study and stratified to receive either ASA alone or ASA plus clopidogrel or ticlopidine. Within each stratum, patients were randomized to receive either low-dose rivaroxaban (2.5 or 5 mg bid) or placebo. 61 In the ATLAS ACS 2 TIMI 51, 15 526 patients were randomized. The mean duration of treatment was 13.1 months. Overall, rivaroxaban significantly reduced the rate of cardiovascular death, MI, and stroke compared to placebo (8.9% vs 10.7%; HR = 0.84; 95% CI 0.74-0.96; P = .008), with a significant reduction seen with both doses of rivaroxaban. For the 5 mg bid dose, this significant reduction was driven by a lower nonfatal MI rate, whereas for the 2.5 mg bid dose a reduction in cardiovascular deaths was the driver. Although an analysis of stent thrombosis was not part of the formal statistical hierarchy, compared with placebo, rivaroxaban reduced the risk of stent thrombosis by 31%. Compared with placebo, rivaroxaban overall was associated with an increased rate of noncoronary artery bypass graft surgery-related major bleeding (2.1% vs 0.6%; HR = 3.96; 95% CI 2.46-6.38; P < .001) and intracranial hemorrhage (0.6% vs 0.2%; HR = 3.28; 95% CI 1.28-8.42; P = .009). However, there was no significant increase in the rates of fatal bleeding with either dose of rivaroxaban compared with placebo (0.3% [overall] vs 0.2%; P = .66).
There are 3 possible reasons why a lower dose of rivaroxaban may have resulted in lower cardiovascular mortality than the higher dose (1) higher rates of bleeding with the 5 mg dose compared with the 2.5 mg dose (fatal bleeding 0.4% [5 mg bid] vs 0.1% [2.5 mg bid]; P = .04) are related to higher mortality rates; 62 (2) bleeding with higher doses may cause intraplaque hemorrhage, making the plaque unstable and causing a fatal MI; (3) when bleeding occurs, often evidence-based treatments like ASA are stopped 63 and mortality may, therefore, increase.
The 2 doses of rivaroxaban studied (2.5 and 5 mg bid) were a quarter to a half of the 20 mg od dose tested in patients with AF. 64 The ATLAS ACS TIMI 46 trial tested doses of rivaroxaban as high as 20 mg daily; however, as mentioned previously, the 2.5 and 5 mg bid doses were selected for the phase III trial based on net clinical outcome (death, MI, stroke, or Thrombolysis In Myocardial Infarction [TIMI] major bleeding). 60 Higher doses of rivaroxaban were associated with increased rates of TIMI major bleeding and bleeding requiring medical attention. In patients with ACS, for whom dual antiplatelet–anticoagulant therapy is proposed to become routine, striking the balance between reduced ischemic events and increased bleeding is particularly important. Although the same principle applies to patients with AF, the 20 mg od dose selected for ROCKET AF was based on phase II trials in the treatment of venous thromboembolism 65 –67 and was subsequently tested in patients for whom dual antiplatelet therapy is not a matter of routine.
Other factor Xa inhibitors have also been evaluated. Darexaban (formerly YM150) is a potent factor Xa inhibitor which has been tested in phase II trials for prevention of venous thromboembolism, 68,69 AF, 70,71 and ACS. 72 In the RUBY-1, 72 higher bleeding rates were observed for all darexaban doses compared with placebo, with no reduction in ischemic events. The development of darexaban has since been stopped. 73 TAK-442 has also been tested in a phase II ACS study, but has yielded disappointing results, with no reduction in major adverse cardiac events. 74
There have also been recent noteworthy trial results for the novel antiplatelet agent vorapaxar, which inhibits thrombin-induced platelet aggregation via the PAR-1 receptor. 75 The addition of vorapaxar to standard therapy in patients with ACS was evaluated in the phase III TRACER trial, in which approximately 97% of the patients were receiving ASA and 87% a thienopyridine at baseline. Follow-up of the trial was stopped early after a median of 502 days on the recommendation of the trial’s Data and Safety Monitoring Board. Although there was no significant difference in the primary efficacy end point (composite of cardiovascular death, MI, stroke, recurrent ischemia with rehospitalization, or urgent revascularization; P = .07), there was a significant reduction in the secondary composite end point of cardiovascular death, MI, or stroke with vorapaxar compared with placebo (14.7% vs 16.4%; HR 0.89; 95% CI 0.81-0.98; P = .02). However, the rate of GUSTO moderate or severe bleeding was significantly increased with vorapaxar versus placebo (7.2% vs 5.2%; HR 1.35; 95% CI 1.16-1.58; P < .001), as was intracranial hemorrhage (1.1.% vs 0.2%; HR 3.39; 95% CI 1.78-6.45; P < .001).
Summary and Conclusions
Current evidence suggests that thrombus formation is mediated by the complex (and simultaneous) interplay of the primary and secondary hemostatic pathways, involving both platelet activation and fibrin formation. However, clinical studies show that a residual risk of adverse cardiovascular events remains with dual antiplatelet therapy, the current standard of care. Therefore, there is both a mechanistic and clinical rationale for targeting the fibrin component of thrombus formation in ACS. The addition of newer anticoagulants to standard of care may help to address the therapeutic shortfall presented by antiplatelets alone, without the drawbacks presented by VKAs. Theoretically, because of their mechanisms of action at the crossover point between the primary and secondary hemostatic pathways, these novel agents have the potential to substantially augment the efficacy of dual antiplatelet therapy. The increased risk of major and intracranial hemorrhage by the addition of factor Xa inhibitors is present throughout the treatment period. This risk of bleeding will be greater in patients who are often excluded from clinical trials, that is the elderly, patients with lower body weight, and patients with reduced renal function. In the real world, the balance of decreased ischemic risk versus increased risk of bleeding may, therefore, be different than that in clinical trials. Future subanalyses may potentially resolve some of these issues. Results seen with the lower rivaroxaban dose in the ATLAS ACS 2 TIMI 51 are encouraging. However, the decrease in ischemic events seen with rivaroxaban on a background of ASA and clopidogrel compared with the standard therapy of ASA and clopidogrel also needs to be validated with other background therapies such as ASA with prasugrel or ASA with ticagrelor.
In addition to ASA plus clopidogrel for secondary prevention in patients with ACS, targeting the coagulation cascade with a factor Xa inhibitor further reduces ischemic events. However, despite positive trial findings, major challenges remain.
Footnotes
Acknowledgments
The author would like to acknowledge Richard Ogilvy-Stewart, who provided medical writing services with funding from Bayer HealthCare Pharmaceuticals and Janssen Research & Development, LLC.
Declaration of Conflicting Interests
The author declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Professor Harvey White has received research grants from the following companies: Sanofi Aventis; Eli Lilly; Medicines Company; NIH; Pfizer; Roche; Johnson & Johnson; Schering Plough; Merck Sharpe & Dohme; AstraZeneca; GlaxoSmithKline; Daiichi Sankyo Pharma Development; and Bristol-Myers Squibb.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.