
Abstract
In spite of treatment with the current standard of care antiplatelet regimens including dual antiplatelet therapy, recurrence rates of ischemic events remain elevated for high-risk patients with atherosclerotic disease. This may be in part attributed to the fact that other key platelet activation pathways remain uninhibited and can thus continue to trigger platelet activation and lead to thrombotic complications. Thrombin is a powerful inducer of platelet activation and mediates its effects directly on platelets through protease activator receptors (PARs), particularly the PAR-1 subtype, making PAR-1 inhibition an attractive approach for reducing atherothrombotic events. These observations have led to the development of several PAR-1 antagonists. Vorapaxar is a direct inhibitor of PAR-1 and the only agent of this class approved for the prevention of recurrent ischemic events in patients with prior myocardial infarction or peripheral artery disease. In the present manuscript, we present a review of the pathophysiologic role of thrombin on thrombotic complications, the impact of vorapaxar on outcomes, including the most recent updates deriving from clinical trials, as well as future perspectives in the field.
Introduction
The key mechanism leading to an acute coronary syndrome (ACS) is based on arterial thrombosis which develops after rupture or erosion of atherosclerotic plaques. 1 The process of platelet activation and aggregation contribute to the development of arterial thrombosis and vascular occlusion. 1 –3 Treatment with antiplatelet agents, therefore, is essential for the treatment and prevention of atherothrombotic complications in patients with ACS. Combination therapy with aspirin and a P2Y12 receptor antagonist, also known as dual antiplatelet therapy (DAPT), has represented the gold standard antiplatelet treatment regimen to prevent recurrences of adverse events in patients with atherothrombotic disease. 4 –6 Currently, 3 oral P2Y12 receptor antagonists are most commonly used and include clopidogrel, prasugrel, and ticagrelor. More potent, prompt, and predictable antiplatelet effects are the main characteristics of prasugrel and ticagrelor compared to clopidogrel, and these drugs are associated with a significant reduction in cardiovascular events in patients with ACS at the expense of higher bleeding risk. 5,6 However, despite the use of standard DAPT regimens, recurrence rate of ischemic events still remains high. This may be in part attributed to the fact that other platelet activation pathways remain uninhibited and thus can continue to induce platelet activation and promote thrombotic complications. 7 –10 These observations underscore the need to identify platelet signaling pathways, other than those blocked by aspirin and P2Y12 receptor inhibitors, to inhibit with the goal of reducing atherothrombotic recurrences. Thrombin is a powerful inducer of platelet activation and mediates its effects directly on platelets through protease activator receptors (PARs), particularly the PAR-1 subtype. 9 Several PAR-1 inhibiting agents have been developed. However, vorapaxar is the only one that has successfully completed phase III clinical investigations and is available for clinical use. 11,12 In particular, vorapaxar is currently approved for the prevention of recurrent ischemic events in patients with prior myocardial infarction (MI) or peripheral artery disease (PAD). 13 The present manuscript will focus on the role of thrombin in thrombus generation, the impact of vorapaxar on outcomes, including most recent updates from clinical trials, as well as future perspectives in the field.
Platelet Activation and Rationale for the Use of Antiplatelet Agents
The generation of platelet-activated thrombosis involves 3 principal steps: (1) an initiation phase involving platelet adhesion, (2) an extension phase that includes activation, additional recruitment, and aggregation of platelets, and (3) a perpetuation phase characterized by platelet stimulation and stabilization of clots. 2,3,14,15 In particular, the initiation phase of hemostasis is started once platelets in the circulation interact with the exposed extracellular matrix (ECM) at the site of vessel injury. The initial “rolling” of platelet at the ECM is mediated by binding between von Willebrand factor immobilized on exposed collagens and the glycoprotein (GP) Ib/V/IX receptor complex located on the platelet surface. This initial interaction in the “rolling” stage is not stable and simply allows the platelet to be in close contact with the ECM of the injured vasculature. Therefore, during this initial phase, platelets can be translocated by the blood stream flow. 15 During this interaction, platelets establish connections between collagen and the immunoglobulin superfamily receptor, GP VI on the platelet surface. Binding of collagen to GPVI generates intracellular signals that change platelet structure to a high-affinity form and lead to the release of secondary adhesion mediators, including thromboxane A2 (TXA2), adenosine diphosphate (ADP), and thrombin. 2,3,15 In addition, injury to the vascular structure also reveals subendothelial tissue factor, and it makes a complex with factor VIIa. Consequently, this complex activates the clotting cascade and leads to the production of thrombin, which further contributes to platelet activation via binding to PAR-1 (Figure 1). 3,16 The released secondary adhesion mediators activate stimulating receptors that is composed of G protein coupled receptors (GPCRs), which contribute to utmost platelet activation. The conversion of platelet GP IIb/IIIa receptor to an active structure is the final process in the process of platelet activation and thrombus formation. The platelet GP IIb/IIIa receptor is known as the main receptor which mediates platelet aggregation and can bind to the extracellular ligands of fibrinogen after conversion to its active form. This process enables platelet–platelet connection, which is the last process of thrombus formation.

Platelet adhesion and aggregation. Platelet adhesion is initially mediated by interactions between the GP Ib/V/IX receptor complex and vWF, enabling subsequent interaction between GP VI and collagen. This induces a high-affinity state of integrins and the release of ADP and TXA2, which bind to the P2Y12 and TP receptors, respectively. Tissue factor locally triggers thrombin formation, which leads to platelet activation via binding to the platelet PAR-1. ADP indicates adenosine diphosphate; GP, glycoprotein; PAR-1, protease activated receptor-1; TP receptor, thromboxane prostanoid receptor; TXA2, thromboxane A2; vWF, von Willebrand factor. Reproduced with permission from Angiolillo et al. 3
The process of platelet activation is complex and various receptors and signaling pathways are involved. Most commonly used oral antiplatelet agents have been developed to block the production of TXA2 (ie, by the cyclooxygenase-1 inhibitor aspirin) and inhibition of the ADP P2Y12 receptor (ie, clopidogrel, prasugrel, and ticagrelor). 17 However, many other signaling pathways remain uninhibited and can thus promote platelet activation and thrombus formation (Figure 2). 9

Platelet activation pathways targeted by current and novel antiplatelet agents. Currently used and emerging antiplatelet regimens are presented. The mechanisms of action of antiplatelet effect are based on blockade of the thromboxane pathway, inhibition of platelet activation by P2Y12 receptor and PAR-1, and serotonin receptor 2A antagonism. *Combined thromboxane receptor antagonists and TXS inhibitors. COX indicates cyclooxygenase; GP, glycoprotein; 5HT2A, serotonin receptor 2A; PAR, protease-activated receptor; TP receptor, thromboxane prostanoid receptor; TXS, thromboxane A2 synthase; vWF, von Willebrand factor. Reproduced with permission from Franchi and Angiolillo. 7
Platelet Activation and Aggregation Mediated by Thrombin
Thrombin is known as a multifunctional serine protease, which plays a critical role in platelet activation. Importantly, the main thrombin source in blood circulation is from the surface of activated platelets. 14,18 During the process of platelet aggregation, thrombin shows its multifunctional properties. First, the conversion of fibrinogen into fibrin is mediated by thrombin, leading to a fibrin-rich clot. Second, thrombin contributes to the activation of platelets by binding to PARs, which are composed of a subset of GPCRs on the platelet surface. 7,9,14,18 There are 4 PAR subtypes which have wide tissue distribution. 19,20 Human platelets express only PAR-1 and PAR-4. 17 Protease activator receptor-1 is considered as the primary thrombin receptor, since it exhibits higher thrombin affinity compared to PAR-4. The biological role of PAR-4 in humans is not fully understood. 21 The first step of PAR-1 activation is initiated after thrombin cleaves its specific site of the N-terminal exodomain on the extracellular portion of the PAR-1, which is followed by binding of the newly exposed N-terminus sequence to the specific site of PAR-1. This binding interaction activates the PAR-1 itself. In particular, activation of the heterotrimeric G proteins (Gα12/13, Gαq, and Gαi/z families) leads to modulation of the various intracellular signaling pathways underlying the effects of thrombin on platelet activation. These include the production of TXA2, release of various substance such as ADP, adrenalin, and serotonin. Protease activator receptor-1 is also expressed by endothelial cell and vascular smooth muscle cells. Therefore, the activation of PAR-1 by thrombin is associated with inflammation, vascular transcriptional activation, vascular smooth muscle cell migration, and proliferation (Figure 3). 9,18,22 Overall, PAR-1 cleavage by thrombin plays a critical role in the process of platelet activation, making PAR-1 inhibition an attractive approach for reducing atherothrombotic events. 23,24

Signaling pathways of PAR-1 activation in platelets. Activation of PAR-1 by binding of the newly exposed N-terminus sequence signal through heterotrimeric G proteins. The initial binding of α-subunits of G12/G13 to ρ GEFs leads to ρ-mediated cytoskeletal responses, result in platelet shape change. The binding of Gαq to phospholipase Cβ leads to calcium mobilization by IP3, activation of PKC, and then secretion of granule. Gαi/z inhibits adenylyl cyclase and consequently decreased cAMP production. PI3K is activated by the Gβγ subunits. Activated PAR-1 can also induce growth-factor “shedding” and activate tyrosine kinases receptor. Consequently, the activation of PAR-1 by thrombin correlated with inflammation, vascular transcripitonal activation, vascular smooth muscle cell migration, and proliferation. cAMP indicates cyclic adenosine monophosphate; DAG, diacylglycerol; GEFs, guanine nucleotide exchange factors; IP3, inositol trisphosphate 3; MAP, mitogen-activated kinase; MLC, myosin light chain; PHD, prolyl hydroxylase domain; PI3K, phosphoinositide-3 kinase; PKC, protein kinase C; SRE, serum response element; WASP, Wiskott-Aldrich syndrome protein. Reproduced with permission from Angiolillo et al. 9
Rationale of PAR-1 Inhibition for Atherosclerotic Disease
In contrast to platelet activation mediated by ADP and TXA2, which are essential for both pathological thrombus formation and routine hemostasis, platelet activation by PAR-1 mainly contributes to pathological thrombus formation. 9 In particular, PAR-1 helps to generate the platelet-rich thrombus, but this cannot spread beyond the initial monolayer of platelets. These findings may explain the role of PAR-1 in pathological thrombus formation, but not in protective hemostasis. 9 In a guinea pig model, PAR-1 blockade contributed to reduction of arterial thrombosis without prolongation of bleeding times or influence of coagulation profiles. 25 In contrast, argatroban which is known as the direct thrombin inhibitor decreased thrombus formation but with significant prolongation in bleeding and coagulation times. 25 In addition, observations from several preclinical investigations demonstrated that conversion of fibrinogen into fibrin by thrombin is more important for hemostasis than platelet activation by PAR-1 activation. 18 Therefore, PAR-1 inhibition would have the theoretical benefit in the prevention of thrombin-mediated platelet activation without changing the function of thrombin in the generation of fibrin, minimizing bleeding complications. 9,18 Moreover, there is a suggested interaction between the PAR-1 and the P2Y12 receptors in the signaling cascade leading to platelet activation, which may create a synergistic effects when these pathways are simultaneously inhibited by specific antagonists. 26,27
A number of PAR-1 antagonists have been developed, but most of them have been limited to preclinical and early phase clinical trials. In 3 phase II studies using atopaxar (Japanese-Lesson from Antagonizing the Cellular Effect of Thrombin, 28 Lesson from Antagonizing the Cellular Effect of Thrombin-ACS, 29 and Lesson from Antagonizing the Cellular Effect of Thrombin-Coronary Artery Disease 30 ), a good safety profile in terms of bleeding with a numerical decrease in the rate of adverse ischemic events was demonstrated. However, atopaxar was shown to be associated with prolongation of corrected QT interval and abnormal liver function. 31 Therefore, the clinical development of atopaxar has been abandoned. Only vorapaxar has completed phase III clinical trial testing and obtained an approval for clinical use. Details on its pharmacology and clinical trial results are elaborated below.
Pharmacologic Profiles of Vorapaxar
Vorapaxar (trade name; Zontivity) is an orally active, small molecule, and a tricyclic 3-phenyl pyridine analog, synthesized based on the natural product of himbacine. 9,24,32 It binds to the PAR-1 in a competitive manner and with high affinity and displays potent inhibitory effects. After oral administration, vorapaxar is rapidly absorbed with over 90% of bioavailability, and it has a dissociation half-life from the PAR-1 of about 20 hours with a long-term half-life (126-269 hours). 24 Although the binding between vorapaxar and PAR-1 is reversible, the long half-life of vorapaxar in plasma makes its effect essentially irreversible, allowing for consistent platelet inhibitory effects. Vorapaxar is metabolized by cytochrome P450 (CYP) 3A4, and the elimination route is mainly feces and secondarily by urine (<5%). 33 The rate of metabolic conversion by the CYP 3A4 enzyme is low, however, coadministration of drugs that interfere with the activity of this enzyme (including ketoconazole, itraconazole, posaconazole, clarithromycin, or rifampin) can affect the pharmacodynamics of vorapaxar. 34 It is also known that the antiplatelet effect of vorapaxar is not affected by hepatic and renal functions. 35,36
Since vorapaxar selectively targets PAR-1 with high affinity (and not circulating thrombin), it blocks thrombin-mediated platelet activation without interfering with thrombin-mediated cleavage of fibrinogen; consequently, the coagulation cascade and bleeding time are not affected. 25 In a recently reported pharmacodynamic study, vorapaxar was shown to prolong occlusion time and shorten lysis time assessed by a point-of-care global thrombosis test, with favorable effects on thrombotic and thrombolytic status. Therefore, it has been suggested that vorapaxar may enhance endogenous thrombolysis, which is frequently impaired in patients with coronary artery disease. 37
Clinical Trials of Vorapaxar
The Thrombin Receptor Antagonist Percutaneous Coronary Intervention (TRA-PCI) trial was performed for the evaluation of safety of vorapaxar. This randomized phase II trial was conducted as a double-blinded and placebo-controlled study in patients undergoing nonurgent PCI or planned PCI (n = 1030). 38 No significantly increased bleeding with vorapaxar compared to placebo was demonstrated. There was also a numerically lower incidence of the composite of death, MI, and stroke (secondary ischemic end point) in patients treated with vorapaxar, without reaching statistical significance. Overall, the result of TRA-PCI suggested a favorable safety and tolerability of vorapaxar, which led to large-scale phase III clinical trials: the Thrombin Receptor Antagonist for Clinical Event Reduction (TRACER) in ACS and Thrombin-Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events (TRA 2P-TIMI 50) as described below.
The TRACER Trial
The aim of the TRACER trial was to define the benefit of adding vorapaxar to standard DAPT on recurrent cardiovascular events in patients with a nonST-segment elevation-ACS (NSTE-ACS; n = 12 944). 39 This randomized trial was conducted in a double blind and placebo-controlled manner in patients who presented an NSTE-ACS within 24 hours. Patients were randomized to vorapaxar and placebo. For the vorapaxar group, a 2.5 mg daily dose of vorapaxar (after a 40 mg loading dose) was received on top of standard antiplatelet regimens, including DAPT with aspirin and clopidogrel. In this trial, the vorapaxar-treated group showed a lower incidence of the primary efficacy end point (a composite of cardiovascular death, MI, stroke, recurrent ischemia with re-hospitalization, or urgent coronary revascularization) compared to the placebo group; however there was no statistically significant difference (vorapaxar 18.5% vs placebo 19.9%, hazard ratio [HR]: 0.92, 95% confidence interval [CI]: 0.85-1.01, P = .07). Notably, vorapaxar demonstrated a significant reduction in the secondary end point, including a composite of cardiovascular death, MI, or stroke (vorapaxar 14.7% vs placebo 16.4%, HR: 0.89, 95% CI: 0.81-0.98, P = .02). This finding is originated from a significant reduction in the incidence of MI. However, the primary safety end point, determined by Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Arteries (GUSTO) severe/moderate bleeding was significantly increased with vorapaxar (7.2% vs 5.2%, HR: 1.35, 95% CI: 1.16-1.58, P < .001). It is important to note that the rate of intracranial hemorrhage (ICH) was 1.1% in vorapaxar-treated patients and 0.2% in placebo (HR: 3.39, 95% CI: 1.78-6.45, P < .001; Table 1). Because of an excess of bleeding, including ICH, the TRACER study was terminated early. Safety concerns emerged particularly for patients who had a history of a prior cerebrovascular event, which also affected the TRA 2P-TIMI 50 trial as described below. Even though study terminated early, this did not affect the outcomes of the trial since the target number for determining efficacy and safety had already been reached. Although the primary objective of the TRACER trial was not met, it provides substantial knowledge on the effects of adding vorapaxar on top of standard antiplatelet therapy in patients with NSTE-ACS.
Efficacy and Safety End points in the TRACER Trial 39 at 2 Years.

Abbreviations: CI, confidence interval; CV, cardiovascular; UCR, Urgent Coronary Revascularization; GUSTO, Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Arteries; MI, myocardial infarction; TIMI, thrombolysis in myocardial infarction; TRACER, Thrombin Receptor Antagonist for Clinical Event Reduction.
The main determinant for the reduced ischemic events with vorapaxar was derived from lowering of MI rates. Vorapaxar showed a 12% risk reduction in first MI, mostly type 1 (HR: 0.88, 95% CI: 0.79-0.98, P = .021) and 14% reduction in total MIs (HR: 0.86, 95% CI: 0.77-0.97, P =.014). 40 In the subgroup of patients treated with PCI (n = 7479, 58% of total number), there were no differences between vorapaxar and placebo in the primary and secondary endpoints.
Patients who underwent the implantation of a bare metal stent tend to receive shorter duration of clopidogrel and showed trends toward higher ischemic benefit from vorapaxar and lower bleeding hazard, compared to patients who underwent the implantation of drug eluting stent. 41 There was no significant difference between groups in the rate of definite or probable stent thrombosis (defined by Academic Research Consortium; vorapaxar 1.7% vs placebo 1.5%). 39 In a subgroup analysis of patients who underwent coronary artery bypass graft (CABG) surgery (n = 1312) during the index hospitalization, there was a remarkable reduction in the primary endpoint with vorapaxar (8.2% vs 12.9%; P = .005), with increased major bleeding which were numerically higher than placebo but did not reach statistical significance. 42 In patients with PAD (n = 936, 7.2% of total population), although there were similar incidences of the composite end point of cardiovascular death, MI, or stroke (21.7% vs 24.8%, P interaction = .787), vorapaxar-treated patients with PAD had lower rates of peripheral revascularization (8.1% vs 9.0%, P = .158) and amputation (0.9% vs 1.5%, P = .107). These effects did not reach statistical significance, but raised interest on the potential value of vorapaxar in patients with PAD. 43 A subgroup analysis based on aspirin dose was performed and showed that patients who were on high-dose aspirin (≥ 300 mg/day) tended to have a worse outcomes in the rate of ischemic and bleeding events, although these differences were not statistically significant. 44 Therefore, in combination with vorapaxar, the use of low-dose aspirin (≤100 mg/day) is recommended. Regarding the coadministration with clopidogrel, there was no interaction between clopidogrel and vorapaxar on outcomes of safety and efficacy. 45
Overall, the results of the TRACER trial demonstrated that, in patients with NSTE-ACS, the addition of vorapaxar to standard DAPT yields only minimal ischemic benefits (not reaching statistical significance for its primary ischemic endpoint), at the cost of significantly increased bleeding, especially ICH. There are some plausible explanations for the negative outcomes of the TRACER trial. In TRACER, about 70% of the study population had no previous MI, nearly 60% of participants were naive to antiplatelet therapy, and only 10% received DAPT before the index hospitalization. The loading doses of clopidogrel (300-600 mg) and vorapaxar (40 mg) would be given to all patients, in addition to other parenteral anticoagulation for the treatment of ACS. Therefore, some of these patients may have been more susceptible to bleeding complications, which is also well known to be associated with an increase in ischemic complications which could have offset the benefits of vorapaxar. This is in contrast with the TRA 2P-TIMI 50 trial in whom patients were first stabilized after their acute event, likely ruling out patients with high bleeding risk. Moreover, patients were not treated with a loading dose of vorapaxar, among other differences as described in details below.
The TRA 2P-TIMI 50 Trial
The TRA 2P-TIMI 50 was a phase III, randomized, double-blinded, placebo-controlled clinical trial designed to evaluate whether intensified antiplatelet therapy by addition of vorapaxar is beneficial for secondary prevention of ischemic events in patients with stable atherosclerotic disease. 46 Patients with a stable atherosclerotic disease manifestations, defined as prior spontaneous MI or ischemic stroke (within the periods of previous 2 weeks to 12 months) or PAD with a history of intermittent claudication (with either an ankle-brachial index <0.85 or history of revascularization for limb ischemia) were enrolled. Patients (n = 26 449) were randomly assigned to receive either 2.5 mg daily dose (without a loading dose) of vorapaxar in addition to standard antiplatelet therapy regimen or placebo. At baseline, aspirin was used in 93.5% of total patients and a thienopyridine was used in 78% of patients with a qualifying diagnosis of MI, in 23.7% of patients of prior stroke, and in 36.8% of PAD. Clopidogrel was the most used; thienopyridine and prasugrel was used only in 0.7% of patients (n = 177) during the study period. 46 The primary end point of this trial was the composite of cardiovascular death, MI, or stroke and the secondary end point was the composite of cardiovascular death, MI, stroke, or recurrence of ischemia driven urgent coronary revascularization. The safety end point was the composite of GUSTO moderate/severe bleeding. However, during the follow-up period of a median of 24 months, a high rate of ICH in patients with a history of stroke in the vorapaxar group was identified by the data and safety monitoring board of trial (as described above, the same as in the TRACER trial), and the discontinuation of vorapaxar for patients with prior stroke was recommended. This policy was also applied for patients with a new stroke event during the study period.
At 3 years, the rate of the primary end point was significantly lower in the vorapaxar-treated group compared with placebo (9.3% vs 10.5%, HR: 0.87, 95% CI: 0.80-0.94, P < .001). There was also a reduction in the secondary end point (11.2% vs 12.4%, HR: 0.88, 95% CI: 0.82-0.95, P = .001; Table 2). The benefit of vorapaxar in the prevention of ischemic events observed in this trial was mainly driven by a significant reduction in the rate of new MI (5.2% vs 6.1%, HR: 0.83, 95% CI: 0.74-0.93, P = .001). Using the universal definition of MI classification system, 47 vorapaxar significantly reduced the incidence of type 1 MI (4.2% vs 4.9% HR: 0.84, 95% CI: 0.73-0.98, P = .024), with a similar effect for type 2 MI (0.6% vs 0.8% HR: 0.74, 95% CI: 0.49-1.10, P = .13). Of note, vorapaxar demonstrated a significant reduction in larger, spontaneous MIs, and a consistent pattern with respect to fatal MI. 48 Importantly, the study population was treated with optimal medical therapy, including aspirin (93.5%), thienopyridine (62.2%), lipid-lowering agent (91.4%), angiotensin-converting enzyme inhibitor/angiotensin receptor blocker (74.0%). 13,46 Therefore, the efficacy of adding vorapaxar for secondary prevention was demonstrated on top of the best contemporary standard of care. However, the rate of the safety end point (GUSTO moderate/severe bleeding) occurred more with vorapaxar compared to placebo (4.2% vs 2.5%, HR: 1.66, 95% CI: 1.43-1.93, P = .001), including a 2-fold increase in ICH (1.0% vs 0.5%, HR: 1.94, 95% CI: 1.39-2.70, P < .001; Table 2).
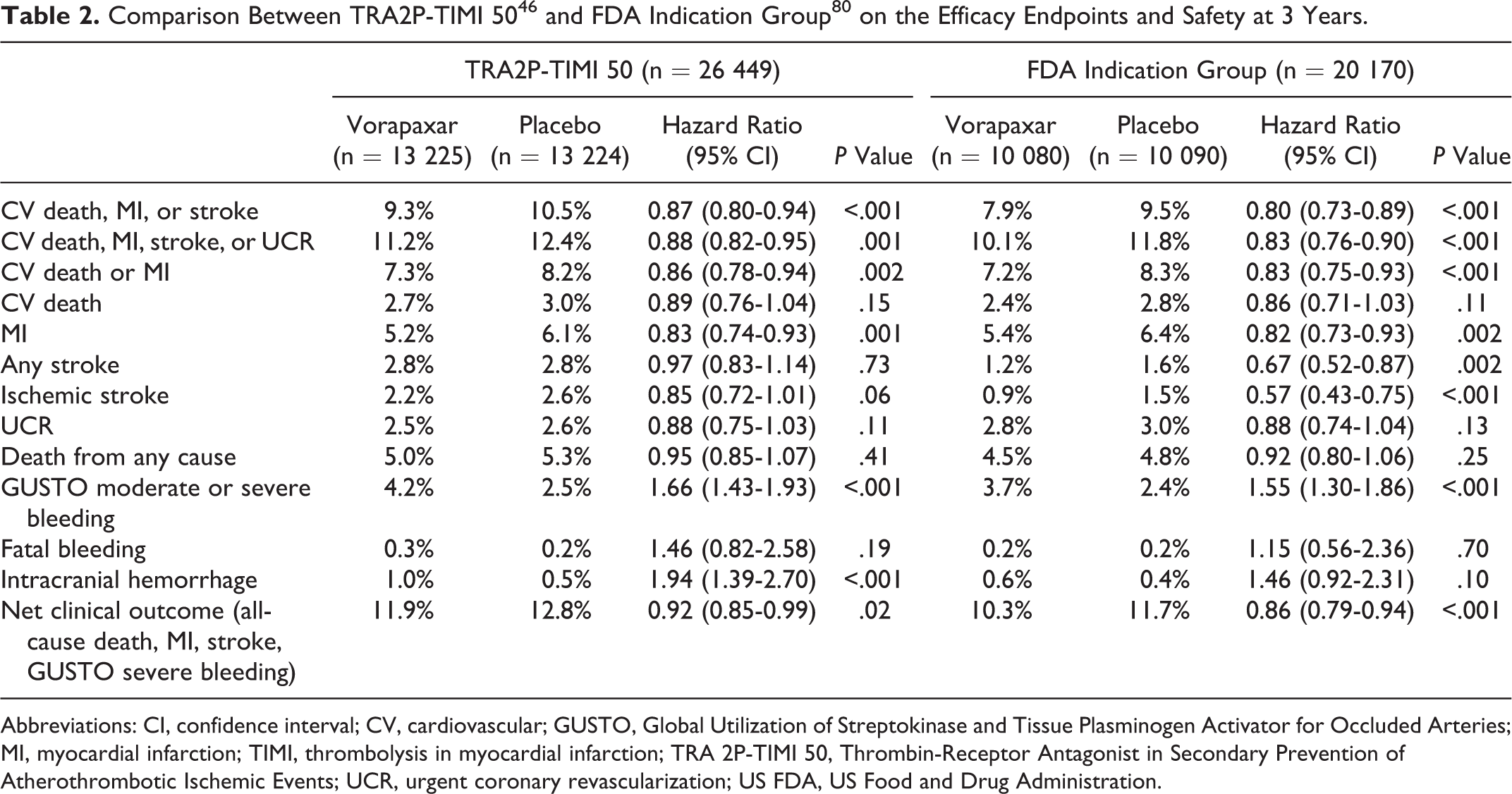
Abbreviations: CI, confidence interval; CV, cardiovascular; GUSTO, Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Arteries; MI, myocardial infarction; TIMI, thrombolysis in myocardial infarction; TRA 2P-TIMI 50, Thrombin-Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events; UCR, urgent coronary revascularization; US FDA, US Food and Drug Administration.
Importantly, in patients with a history of stroke, major bleeding and ICH were significantly increased with vorapaxar, without any improvement in ischemic events. Although there was a significant interaction in a subgroup analysis of patients with low body weight (LBW, <60 kg), vorapaxar did not provide a beneficial effect on the study outcome in patients of LBW (P = .03 for interaction). Notably, vorapaxar was found to significantly reduce the hazard of adverse events (such as cardiovascular death, MI, or stroke) in patients whose qualifying diagnosis for enrollment was MI, but not in patients whose qualifying diagnosis was stroke or PAD (details described below).
Overall, the results of TRA 2P-TIMI 50 showed that inhibition of the PAR-1 pathway of platelet activation in addition to standard antiplatelet therapy is effective in the secondary prevention of atherothrombotic events, with the exception of patients with a previous stroke, at the expense of bleeding complication. The role of vorapaxar in the secondary prevention of ischemic events is more clearly understood by several post hoc and subgroup analysis of the TRA 2P-TIMI 50 trial (Figure 4).

Efficacy and safety of vorapaxar in major subgroups of the TRA 2P-TIMI 50 trial. (A) TRA 2P-TIMI 50 total population 45 (n = 26 449); (B) patients with prior ischemic stroke 49 (n = 4883); (C) patients with prior MI 48 (n = 17 789); (D) patients with prior MI and low risk of bleeding 48 (defined as age ≤75 years, no history of CVA, and body weight ≥60 kg; n = 14 909); (E) patients with PAD 56 (n = 3787); (F) patients with DM and prior MI without stroke 64 (n = 3623); (G) patients with prior MI or PAD and no history of TIA/stroke 79 (US FDA-approved population, n = 20 170). Data presented as hazard ratios (95% confidence interval) for vorapaxar versus placebo. CVA indicates cerebrovascular accident; DM, diabetes mellitus; MI, myocardial infarction; PAD, peripheral vascular disease; TIA, transient ischemic attack; TRA 2P-TIMI 50, Thrombin-Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events; US FDA, US Food and Drug Administration.
Subgroup Analysis of the TRA 2P-TIMI 50 Trial According to Qualifying Study Entry Criteria
Patients With a Prior MI
The largest prespecified subgroup analysis was represented by patients with a qualifying diagnosis of prior MI (n = 17 779; 67% of total patients), and most of the benefit from vorapaxar was proved in this subgroup, as compared with patients with stroke history or PAD (P = .058, for interaction). 49 Vorapaxar significantly reduced the primary end point in this subgroup (8.1% vs 9.7%, HR: 0.80, 95% CI: 0.72-0.89, P < .0001). This finding was consistent across types of MI (ST-segment elevation MI, nonST-segment elevation MI), timing from qualifying MI to randomization (<3 months, 3-6 months or >6 months), as well as according to usage of thienopyridine. 49 However, the incidence of GUSTO moderate or severe bleeding was more frequent with vorapaxar than placebo (3.4% vs 2.1%, P < .0001), as well as TIMI non-CABG major and TIMI clinically significant bleeding. Vorapaxar was also associated with a numerical, statistically nonsignificant, increase in fatal bleeding (0.2% vs 0.1%, P = .3) and in ICH (0.6% vs 0.4%, P = .076). Importantly, vorapaxar demonstrated a significantly better net clinical outcome (a composite of cardiovascular death, MI, stroke, urgent coronary revascularization, and GUSTO moderate or severe bleeding compared to placebo (12.5% vs 13.4%, P = .038). A post hoc outcome analysis was also conducted in patients with MI at low risk of bleeding (n = 14 909, 84% of prior MI population), which excluded patients >75 years of age, < 60 kg of body weight, and history of transient ischemic attack (TIA)/stroke. 49 In this subgroup analysis, vorapaxar significantly reduced the rate of the primary end point compared with placebo (6.8% vs 8.6%, HR: 0.75, 95% CI: 0.66-0.85, P < .0001). Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Arteries moderate or severe bleeding was also lower than in the analysis of the entire population, although it was still significantly higher with vorapaxar than placebo (2.7% vs 1.8%, HR: 1.52, 95% CI: 1.20-1.93, P = .0006). The incidence of ICH in this subgroup showed similar patterns without reaching statistical significance (0.5% vs 0.4%, HR: 1.39, 95% CI: 0.79-2.43, P = 0.25). 49
Patients With a History of Stroke
A total of 4 883 patients in the TRA 2-TIMI 50 trial were enrolled with a qualifying diagnosis of stroke (ischemic stroke in the prior 2 weeks to 12 months). In this subgroup, vorapaxar was associated with a significantly increased risk of ICH (2.5% vs 1.0%, P < .001), and GUSTO moderate or severe bleeding (4.2% vs 2.4%, P <.001), compared with placebo. Despite of a remarkable increase in bleeding complications, vorapaxar did not present any additional benefit in the primary ischemic end point (13.0% with vorapaxar vs 11.7% with placebo, P = .75), as well as ischemic stroke (8.6% with vorapaxar vs 7.1% with placebo; P = .90). 50 In addition, the risk of ICH also increased with vorapaxar in the 498 patients of prior TIA without a known prior stroke who qualified as MI or PAD subgroup, but not reaching the statistical significance (1.9% for vorapaxar vs 0.5% for placebo, P = .23, P for interaction = .40). 50
However, it is important to note that in patients with prior MI or PAD without prior TIA/stroke history (n = 20 170, 76.3% of total population), vorapaxar significantly reduced the occurrence of first ischemic stroke (HR: 0.57, 95% CI: 0.43-0.75, P < .001). 51 In patients who experienced a new ischemic stroke, vorapaxar did not increase the rate of hemorrhagic conversion after stroke (HR: 1.19, 95% CI: 0.49-2.91, P = .70). Although the incidence of hemorrhagic stroke was increased with vorapaxar (HR: 2.79, 95% CI: 1.00-7.73, P = .049), vorapaxar reduced the total incidence of stroke in patients with prior MI or PAD without a prior TIA/stroke (HR: 0.67, 95% CI: 0.52-0.87, P = .002). 51 The different influence of vorapaxar in the prevention of new ischemic stroke events among patients with or without a prior cerebrovascular accident (CVA) might be in part originated from stroke etiology. Classification of the ischemic stroke type indicates the qualifying stroke to be large vessel in 35%, small vessel (lacunar) in 47%, and other or unknown in the remainder. 50 Therefore, lacunar stroke was the majority of patients enrolled as a qualifying diagnosis of stroke in the TRA 2P-TIMI 50 trial. Outcomes with intensive antiplatelet therapy appear to differ among subsets of stroke patients, including harm in patients with lacunar stroke, and benefit in acute ischemic stroke. 52 –55 There are suggestions that poor outcome in the previous ischemic cohort may be originated from the high proportion of lacunar stroke in the TRA 2P-TIMI 50 trial. 56 However, subgroup analysis based on the type of qualifying ischemic stroke did not show any statistically significant differences in the effect of vorapaxar. 50
In summary, adding vorapaxar to antiplatelet therapy patients with prior stroke or TIA increased the risk of bleeding, including ICH, without any ischemic benefit including ischemic stroke, making prior CVA an absolute contraindication for the use of vorapaxar.
Patients With a History of PAD
In the TRA 2P-TIMI 50 trial, a total of 3787 patients (14.3% of study population) enrolled with a qualifying diagnosis of PAD. Although the overall effect of vorapaxar in this subgroup was consistent with the whole study population (P = 0.35 for interaction), treatment with vorapaxar did not show any significant reduction in the risk of the primary efficacy end points compared with placebo (11.3% vs 11.9%, HR: 0.94, 95% CI: 0.78-1.14, P = .53) and GUSTO moderate or severe bleeding significantly increased (7.4% vs 4.5%, P = .001). However, vorapaxar significantly reduced the incidence of hospitalization for acute limb ischemia (ALI; 2.3% vs 3.9%, P = .006) and all revascularization procedures for PAD (18.4% vs 22.2%, P = .017). 57
The impact of vorapaxar was similar across all PAD types, including the thrombotic complication of bypass graft and in situ thrombosis of native vessel. 58 Findings of a favorable effect on graft thrombosis are particularly interesting because previous trials of thienopyridines and anticoagulants have not shown consistent benefit for graft patency in trials which were conducted after bypass surgery. 59 –61 Another substudy from TRA 2P-TIMI 50 is for the analysis of patients (n = 5 845 patients) who had a known history of PAD at randomization regardless of how they qualified for the trial. Compared with placebo, there was also significant reduction in peripheral revascularization (mainly from reduction of surgical revascularization) among patients treated with vorapaxar. 62
Because of the favorable effect on all peripheral revascularizations and graft patency, the potential for “extra-platelet effects” of vorapaxar on atherosclerosis progression has been raised. As described above, PAR-1 is distributed in various cell types, including endothelial and smooth muscle cells. Since its activation by thrombin in these cells have mitogenic effects, vorapaxar may reduce the progression of atherosclerosis with vessel remodeling, thus preventing vessel luminal narrowing. 18,63 Further dedicated studies are needed to support a specific role of vorapaxar for patients with PAD. So far, few medical therapies have been shown to reduce the rate of ALI and peripheral artery revascularization in large prospective, randomized trials 64 ; therefore, vorapaxar will be an important option for the medical treatment of these patients.
Additional Cohort Analysis
Patients With Diabetes Mellitus
The efficacy of vorapaxar in patients with diabetes mellitus (DM) was evaluated in a subgroup analysis composed of 16 896 patients with prior MI without a history of TIA/stroke. The primary end point was nearly 2-fold higher in patients with DM than in patients without DM; the event rates were 14.3% in DM and 7.6% in non-DM group in the analysis of the placebo group. In patients with DM (n = 3623, 21% of this subgroup), vorapaxar significantly reduced the composite of cardiovascular death, MI, or stroke (11.4% vs 14.3%, HR: 0.73, 95% CI: 0.60-0.89, P = .002). Similar to other subgroup analysis, vorapaxar significantly increased the rate of GUSTO moderate/severe bleeding in patients with DM (4.4% vs 2.6%). The effect of vorapaxar was consistent in reduction of the composite of cardiovascular death, MI, or stroke among patients without DM (6.3% vs 7.6%, HR: 0.81, 95% CI: 0.71-0.93, P = .003, P = .40 for interaction). 65 However, greater absolute risk reduction (ARR) was achieved with vorapaxar in patients with DM (−3.50% of an ARR) than in patients without DM (3.50% vs 1.36%). Consequently, the number needed to treat was 29 in patients with DM compared to 74 in patients without DM.
The reasons for the enhanced benefit of vorapaxar in patients with DM may be attributed to multiple factors. First, the inherent increased risk of atherothrombotic complications among patients with DM allows us to observe a treatment effect of greater magnitude. 65 Second, patients with DM are distinguished by their heightened platelet reactivity and decreased response to standard antiplatelet regimens. 66 –69 Patients with diabetes in fact have increased platelet turnover rates which lead to the introduction into circulation of hyperreactive platelets which are no longer inhibited by agents, such as aspirin, that have short systemic half-lives. 70 This can lead to the elevated level of TXA2 biosynthesis. 71 Diabetes mellitus platelets are also characterized by impaired clopidogrel metabolism leading to lower active metabolite levels with increased platelet reactivity compared to patients without DM. 66,72 Upregulation of platelet signaling pathways, including PAR-1, in DM platelets may therefore make them more responsive to the effects of vorapaxar. 66 Therefore, these observation suggest diabetic patients with a prior MI to represent a target population to consider the use of vorapaxar. Further insights on the functional impact of vorapaxar in patients with DM is being explored in the ongoing Optimizing anti-Platelet Therapy In DM (OPTIMUS)-5 trial (NCT 02548650).
Patients With CABG
Among patients qualified on the basis of MI or PAD without history of TIA/stroke, a total of 2942 patients had a history of CABG prior to randomization and 367 patients underwent CABG during the investigation. 73 In the group of patients with a prior CABG, vorapaxar significantly reduced the risk of cardiovascular death, MI, or stroke (11.9% vs 15.6%, HR: 0.71, 95% CI: 0.58-0.88, P = .001). Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Arteries moderate/severe bleeding was significantly increased with vorapaxar in patients with a prior CABG (HR: 1.87, 95% CI: 1.28-2.72, P = .001). However, the increased number of bleeding events was driven largely by an increase in GUSTO moderate bleeding (5.1% vs 2.0%, P < .001) rather than GUSTO severe bleeding (1.8% vs 1.7%, P = .72). In addition, the incidence of TIMI CABG major bleeding was nonsignificantly increased in patients undergoing CABG while receiving vorapaxar (6.3% vs 4.1%, HR: 1.53, 95% CI: 0.58-4.01, P = .39). However, there was no increase in fatal bleeding, reoperations to control bleeding, or ICH in patients with CABG who received vorapaxar within the 7 days preceding operation. 73
Overall, considering the favorable outcomes in both TRACER and TRA 2P-TIMI50, vorapaxar may play a role in antiplatelet therapy of patients with prior CABG or before CABG. Patients treated with CABG are at high risk for recurrence of thrombotic adverse events, including graft failure and progression of native vessel disease, and thus have the potential to obtain particular benefit from intense antiplatelet therapy. Since thrombin generation is increased both during and after CABG, vorapaxar has the ability to reduce recurrent atherothrombotic events in patients who have undergone CABG. 74 Further dedicated investigations are needed to support the clinical utility of vorapaxar in patients undergoing CABG.
Patients Who Experienced a New ACS
It is uncertain for clinicians whether the administration of vorapaxar in patients who experience a new ACS while receiving vorapaxar should be continued, because in the higher bleeding event rates demonstrated in the TRACER trial when vorapaxar was initiated in an NSTE-ACS setting. During the study follow-up of patients (n = 20 170) without history of CVA, 1712 patients experienced a newly developed ACS event (799 patients in the vorapaxar group and 913 in placebo). 75 Within 7 days of an ACS, the incidence of GUSTO severe bleeding was not different between patients with vorapaxar and placebo (0.8% vs 0.8%) and GUSTO moderate/severe bleeding was slightly increased with vorapaxar compared to placebo (2.5% vs 1.6%, HR: 1.59, 95% CI: 0.78-3.24, P = .21). The rate of cardiovascular death, MI, or stroke in the 7 days after a new ACS was lower in patients allocated to vorapaxar compared to placebo (2.4% vs 4.4%, HR: 0.54, 95% CI: 0.31-0.93, P = .027). 75 Therefore, in clinical practice, it is reasonable to continue vorapaxar if a new ACS occurs to patients while receiving it.
Stent Thrombosis Analysis
For the analysis of vorapaxar effect on stent thrombosis rates, patients (n = 14 042, 53% of the entire study population) who had a history of PCI with coronary stents before study enrollment and 449 patients who additionally received a PCI with coronary stent during the trial were selected. During the study follow-up periods, a total of 152 (1.4%) definite stent thrombosis occurred, and these events were mostly late or very late (92%). The rate of definite stent thrombosis was significantly reduced in the vorapaxar group compared to placebo (1.1 vs 1.4%, P = .037). This effect of vorapaxar on decreasing stent thrombosis was consistent, irrespective of time from stent implantation, DM history, usage of DES, and medication of DAPT at randomization. 76 The mechanism of vorapaxar in the prevention of stent thrombosis is not clear, however, it might be originated from a possible role of inhibition of PAR-1 in the vascular smooth muscle and endothelium.
Analysis According to Background Antiplatelet Therapy
In the subgroup analysis including 16 897 patients who qualified with a previous MI without a history of TIA/stroke, the reduction in thrombotic events with vorapaxar was highly consistent whether patients were managed with or without a thienopyridine. Although bleeding was increased with vorapaxar, the relative risk of significant bleeding events was not altered by overlap with a thienopyridine, and net clinical outcome was improved with vorapaxar in patients who were also treated with clopidogrel. 77
In another post hoc analysis, Scirica et al evaluated the impact of aspirin dosing (<100 mg in 6988 patients, 100-162 mg in 7704 patients, >162 mg in 2755 patents) on the effects of vorapaxar. 78 In most of patients (84%) who received aspirin in TRA 2P-TIMI 50 trial, the dose was ≤162 mg daily. The relative risk of bleeding was not higher in vorapaxar-treated patients while on high-dose aspirin. However, since most patients were treated with low dose (≤162 mg per day), the risk of bleeding with high-dose aspirin in combination of vorapaxar could have been underestimated. Overall, results from subgroup analyses of both TRACER and TRA 2P-TIMI50 support the use of a low-dose regimen of aspirin in patients treated with vorapaxar. 45
Practical Recommendations for the Use of Vorapaxar
Given the beneficial effect observed in patients with prior MI and PAD in the TRA 2P-TIMI50 trial, in May 2014 the US Food and Drug Administration (FDA) approved vorapaxar for clinical use. In particular, the FDA indication for vorapaxar use is for the reduction of thrombotic cardiovascular events in patients with a prior MI or PAD. Vorapaxar is contraindicated in patients with a history of stroke, TIA, or ICH. Vorapaxar should be used with precaution in patients who have bleeding risk factors, including old age (>75 years), LBW (<60 kg), history of bleeding disorders, impaired hepatic function, renal dysfunction, and in patients using certain concomitant medications (eg, anticoagulants, fibrinolytic therapy, chronic nonsteroidal anti-inflammatory drugs, selective serotonin reuptake inhibitors, serotonin norepinephrine reuptake inhibitors). In particular, anticoagulants such as warfarin and strong inhibitors or inducers of CYP3A enzyme should not be used concomitantly. Vorapaxar should be administered as an add-on therapy to the standard of care antiplatelet drugs being used. The European Medicines Agency (EMA) recommendations for vorapaxar use are similar to those provided by the FDA. 79 The 2 agencies differ with regard to the following: first, the EMA specify the indication for patients with PAD as “symptomatic PAD,” while to the FDA refers to simply “PAD”; second, the EMA specifies the time frame for initiating vorapaxar which is in line with the TRA-2P trial design (should be started 2 weeks after an acute event and if possible the first 1 year from the MI event), while the FDA does not provide these specifications; third, severe hepatic impairment is a contraindication per the EMA.
In the subgroup analysis of the FDA-approved patient population, which included the cohort of participants with a prior MI or PAD without history of TIA/stroke (n = 20 170; 76% of the overall trial population). 80 Patients receiving vorapaxar had a lower rate of the primary endpoint compared with placebo (7.9% vs 9.5%, HR: 0.80, 95% CI: 0.73-0.89, P < .001), as well an increased rate of bleeding events (3.7% vs 2.4%, HR: 1.55, 95% CI: 1.30-1.86, P < .001). Importantly, in this subgroup population, vorapaxar did not increase the rate of ICH (0.6% vs 0.4%, P = .10) or fatal bleeding (0.2% vs 0.2%, P = .70; Table 2).
Recently, a risk score was developed from the cohort of patients with a prior MI who were randomized to the placebo group in the TRA 2P-TIMI 50 trial (n = 8598) in which 9 clinical risk indicators (age ≥75 years, DM, hypertension, smoking, PAD, previous stroke, previous CABG, history of heart failure, and renal dysfunction) were identified. 81 These risk indicators are useful in selecting high-risk patients and demonstrate a pattern of increasing absolute benefit from treatment with vorapaxar. The greater absolute risk according to the sum of risk indicators led to proportionately greater ARRs (Figure 5). 81 In particular, patients in the lower risk group present no ARR of cardiovascular events (3-years Kaplan-Meier rate, 3.5% for vorapaxar vs 3.6% for placebo). In contrast, in the intermediate risk group there was an ARR of 2.1% (6.0% vs 8.1%) with a number needed to treat (NNT) of 48 to prevent 1 cardiovascular event, whereas there was a 3.2% ARR (14.5% vs 17.7%) with an NNT of 31 in the high-risk group. It is reasonable to remember that the benefit of vorapaxar regarding the prevention of ischemic event will be maximized when it is used in high-risk patients.

Kaplan-Meier curves for efficacy, safety, and net clinical outcomes by risk category (in patients with prior MI and no history of TIA/stroke). (A) Cumulative incidence of efficacy and safety endpoints by treatment with vorapaxar or placebo in the patients of low risk category, (B) in the patients of intermediate risk category, (C) in the patients of high-risk category patients, (D) net clinical outcome of all-cause mortality, MI, stroke, or GUSTO severe bleeding by risk category and vorapaxar. P for trend <.0001 across risk categories for all end points except GUSTO severe bleeding, for which P for trend <.01. P for interaction for cardiovascular disease/MI/ischemic stroke = 0.38, GUSTO severe bleeding = 0.13, and all-cause mortality, MI, stroke, or GUSTO severe bleeding = 0.24. ARI indicates absolute risk increase; ARR, absolute risk reduction; GUSTO, global use of strategies to open occluded coronary arteries; ITT, intention to treat; MI, myocardial infarction; NNT, number needed to treat; TIA, transient ischemic attack. Reproduced with permission from Bohula et al. 81
Future Perspectives
The proven efficacy of vorapaxar has been evaluated as an add-on therapy in patients already on standard of care including aspirin and clopidogrel. This approach however is associated with a higher rate of bleeding complication and often not appealing for patients who would thus be on triple antiplatelet therapy. Therefore, strategies are necessary to minimize the risk of bleeding complications when using vorapaxar as well as enhancing ease of use. Thus, studies are needed to assess vorapaxar as an add-on therapy to a single antiplatelet agent or even as monotherapy. Recently, there has been emerging interest in studying novel antithrombotic agents in addition to a P2Y12 receptor inhibitor, and withdrawing aspirin. The rationale for this is that aspirin is associated with an increased risk of gastrointestinal bleeding complications. 82 The ongoing OPTIMUS-5 study is assessing the pharmacodynamics effects of vorapaxar in both patients with DM and without DM on background of aspirin and clopidogrel therapy and assessing the impact of withdrawal of aspirin therapy in these participants. There is also a need to understand the effects of vorapaxar in patients treated with the newer generation P2Y12 receptor inhibitors, prasugrel and ticagrelor. In fact, most patients on P2Y12 receptor inhibiting therapy in phase III clinical testing with vorapaxar were on clopidogrel. However, in current practice many patients with post-MI are being treated with prasugrel or ticagrelor. Importantly, these drugs have been suggested to modulate platelet signaling pathways other than P2Y12 and that aspirin may offer limited additive effects in the presence of potent P2Y12 receptor blockade. 83,84 Therefore, the role of vorapaxar as a component of a dual antiplatelet treatment regimen (in combination with a novel potent P2Y12 receptor blocker and stopping aspirin) represents another area of interest, which is currently being investigated in the ongoing adjunctive VORApaxar therapy in patients with prior MI treated with new generation P2Y12 Receptor Inhibitors PRAsugrel and TICagrelor pharmacodynamic investigation (NCT 02545933). At this time, there are no large scale studies planned to assess the clinical effects of the above mentioned strategies with vorapaxar.
Conclusion
Although the use of vorapaxar did not show a significant benefit in patients enrolled in the acute phase of an NSTE-ACS, the results of TRA 2P-TIMI 50 trials demonstrated that inhibition of PAR-1 induced platelet activation with vorapaxar in addition to standard antiplatelet therapy is effective for the prevention of recurrent thrombotic events, particularly in patients with previous MI, at the cost of increased bleeding. Certain patient cohorts achieved more benefit than others with adjunctive treatment with vorapaxar, while in some patients there was harm. Therefore, appropriate patient selection is pivotal when deciding to initiate vorapaxar therapy. Future investigations are indeed warranted to better define the efficacy and safety profile of vorapaxar in the current era of pharmacological approaches of high-risk patients with atherosclerotic disease manifestations.
Footnotes
Authors Contributions
J. Moon and D. Angiolillo contributed to conception, design, acquisition, and interpretation and drafted the manuscript. F. Franchi and F. Rollini critically revised the manuscript and gave final approval. All authors agree to be accountable for all aspects of work ensuring integrity and accuracy
Declaration of Conflicting Interests
Dr Angiolillo reports receiving payments as an individual for: (a) consulting fee or honorarium from Amgen, Bayer, Sanofi, Eli Lilly, Daiichi-Sankyo, The Medicines Company, AstraZeneca, Merck, Chiesi, Pfizer, and PLx Pharma; (b) participation in review activities from Johnson & Johnson, and St Jude Medical. Institutional payments for grants from Amgen, Glaxo-Smith-Kline, Eli Lilly, Daiichi-Sankyo, The Medicines Company, AstraZeneca, Janssen Pharmaceuticals, Inc, Osprey Medical, Inc, Novartis, CSL Behring, and Gilead.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.