
Abstract
While blood platelets express several G-protein-coupled receptors (GPCRs) that play pivotal roles in their activation, several diseases, for example thrombotic disorders, may develop if these receptors are inappropriately activated. Thus, these receptors have been the subject of investigations to design therapeutic interventions for managing multiple thrombosis-based disease states. One such GPCR, the thromboxane A2 receptor (TPR), remains resistant to such interventions. The present review provides a critical examination of the binding, structural biology, and signaling of TPRs. The review also provides a rationale for using principles of “drug rediscovery” as an alternative/viable approach for the therapeutic targeting of TPRs. To this end, it is noteworthy that many US Food and Drug Administration (FDA)–approved drugs have been found to selectively (and nonselectively) block TPR-mediated functional responses, for example platelet aggregation, as described in this review. Therefore, while none of the antagonists, thus far developed for targeting TPRs, have made it into clinical use, this peculiar receptor can be antagonized by a large number of drugs used for indications unrelated to thrombosis.
Introduction
Platelet activation is a complex process involving a host of independently stimulated, yet redundantly mediated pathways of activation. 1 –6 One such pathway is triggered by an arachidonic acid (AA) derivative, the eicosanoid thromboxane A2 (TXA2), through binding 7 –9 to a receptor protein known as the thromboxane/prostaglandin H2 (PGH2) receptor (abbreviated as TPR). Thromboxane/prostaglandin receptor is widely distributed in different organ systems and is localized on both cell membranes and intracellular structures. 10 The initial purification and cloning of this receptor from human placenta established this protein as a member of the G-protein-coupled receptor (GPCR) superfamily. Thromboxane/prostaglandin receptor consists of 7 transmembrane™ spanning regions, 3 extracellular loops (ELs), and 3 intracellular loops ([iLs] see Figure 1). This receptor is not only found on platelets but also on macrophages, monocytes, vascular endothelial cells, and smooth muscle cells. Thromboxane/prostaglandin receptor can recognize other endogenous ligands such as the endoperoxide PGG2 11 and isoprostanes. 12
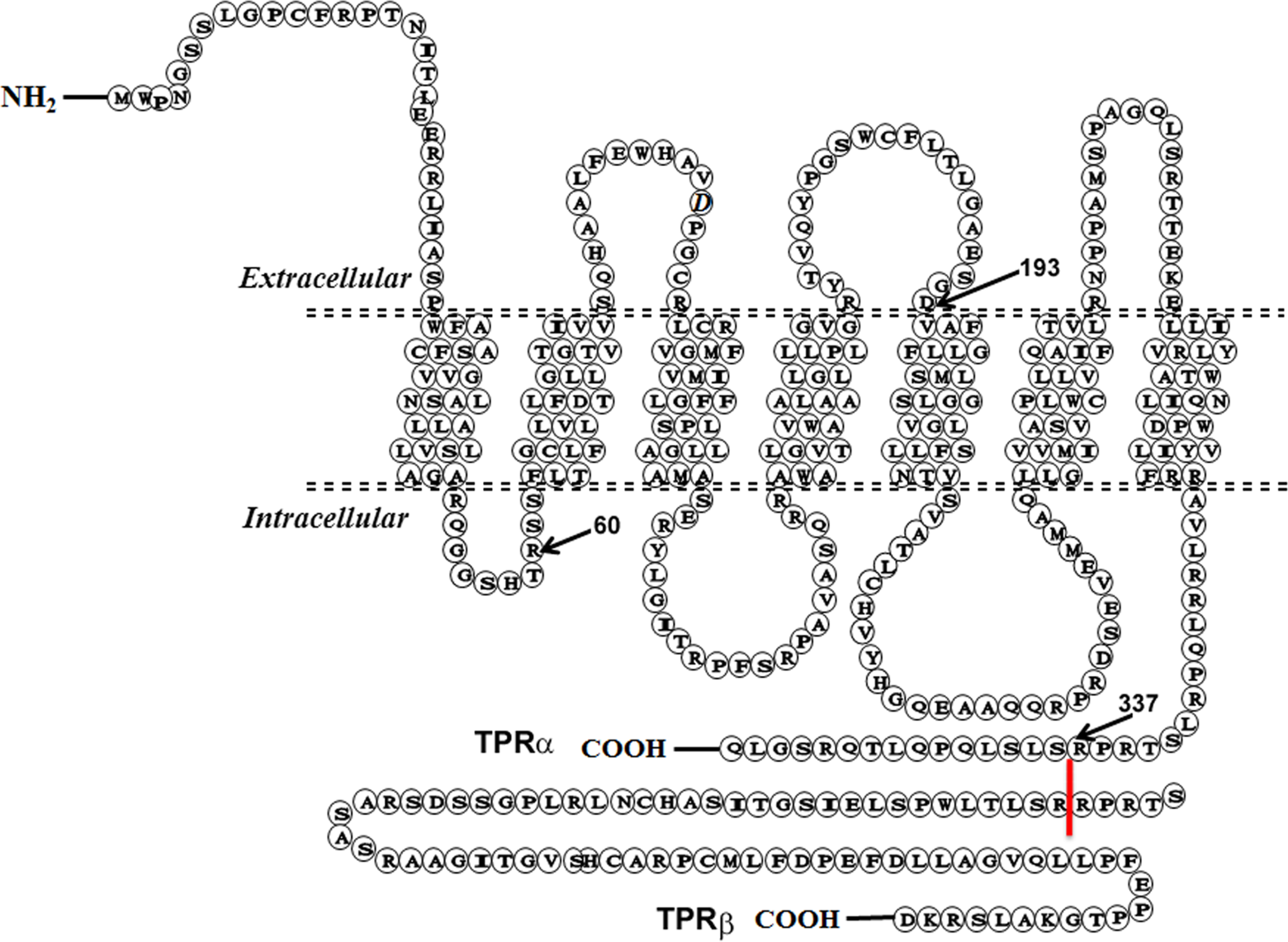
The 2-dimensional topology of the human thromboxane A2 receptor. The thromboxane A2 receptor exists in 2 isoforms, TPR-α and TPR-β, of which only the β isoform differs at the C-terminal tail within the intracellular space. The figure depicts the α isoform with its 3 distinct extra- and intracellular loops and its 7 transmembrane (TM) regions. The C-terminal tail of the β isoform is shown beginning at the 338th amino acid position. 54 × 39mm (300 × 300 DPI).
Platelets are known to be a principal producer of TXA2, which upon binding to TPR initiates signaling cascades that regulate alterations in the cytoskeleton, platelet adhesion, vesicle trafficking, and other platelet activation responses. Thromboxane/prostaglandin receptor has been shown to regulate a host of effectors, including phospholipase C (PLC), several small guanosine triphosphate hydrolases (GTPases), and adenylyl cyclase (AC). 13 Thus, the TXA2 pathway plays an essential role 14 –16 in hemostasis, and in the case of inappropriate activation, the development of thrombotic/occlusive disorders. This makes it an important target for therapeutic interventions.
The principal strategies for targeting the TXA2 pathway for therapeutic purposes to date have focused on either modulating the synthesis of TXA2 17,18 or interfering with its receptor binding. While the former has been successfully targeted by the platelet cyclooxygenase 1 (COX-1) inhibitor aspirin, this therapeutic agent is associated with undesirable adverse effects 19,20 and resistance. 21 Moreover, while TPR antagonists have been successfully developed, 22 –25 none is currently available for clinical use. Thus, there remains considerable interest in the development of TPR antagonists. The focus of this review is to describe the biology, structural features, mechanisms of action and drug interactions with TPR and to provide a foundation for rational theoretical design of novel TPR antagonists.
Structural Features of the Thromboxane A2 Receptor
A comprehensive structural analysis of TPR-binding domains is a logical step in identifying or designing novel TPR antagonists. In this connection, a single gene on chromosome 19p13.3 leads to the expression of 2 (Figure 1) separate TPR isoforms. 24 One is the originally cloned TPR from human placenta, consisting of a primary structure of 343 amino acids and known as the “α” isoform. 26 The other is a splice variant cloned from the endothelium having a primary structure of 407 amino acids and called the “β” isoform. 24 Comparison of the 2 sequences reveals that even though the first 328 amino acids are the same for both isoforms, the β isoform exhibits an extended C-terminal cytoplasmic domain. 26 –28 This variation is due to alternative splicing within exon 3 of the TPR gene. 27 Furthermore, the expression of each protein is not equal within or across different cell types. Thus, while platelets express high concentrations of the α isoform, expression of the β isoform protein has not been documented (ie, platelets thought to possess only residual ribonucleic acid (RNA) for the β isoform). 29
Several studies have focused on elucidating the key domains and specific amino acid residues that coordinate ligand binding to TPR. 30,31 An early finding in the GPCR bovine rhodopsin revealed a putative binding domain in its seventh TM region (TM7). 32 Analogously, the focus of initial studies was the TM regions of the TPR protein. By introducing point mutations to TM region amino acids or by constructing TM chimeras, the ligand-binding activity of TPR was reduced. Ultimately, all regions but TM2 were implicated in mediating TPR-ligand binding. 33 –37
However, other studies suggest the involvement of different TPR regions in ligand coordination. 38 Experiments employing a biotinylated TPR antagonist 38 suggest that ligands interact with extracellular-binding domains. A possible explanation for this discrepancy involves the loss of ligand binding following the introduction of point mutations in the TM regions. Thus, these point mutations appear to result in alterations in TPR that do not directly involve the binding site but rather receptor structural conformation.
An additional study using photoaffinity-labeling and site-specific antibodies demonstrated that the C-terminal segment of the second extracellular loop (C-EL2) of TPR, especially the region of Cys183 to Asp193, was an important region for ligand binding. 39 Later studies revealed the importance of specific residues in the C-EL2 of TPR. For example, a nuclear magnetic resonance (NMR) study revealed that Val176, Leu185, Thr186, and Leu187 were possible residues for ligand binding. 31 Additionally, separate mutagenesis work revealed that differences exist in the amino acids needed to coordinate agonist versus antagonist binding. 40 It was found that Asp193 is a key amino acid residue for both 40 whereas Phe184, Thr186, and Ser191 are important residues only for antagonist binding. This is an important distinction, suggesting the need for careful examination of key pharmacophores of endogenous or artificial agonists in order to rationally develop TPR antagonists.
In addition to the need for a competent ligand-binding domain, TPR must also remain competent to transduce its signal to downstream messengers and effectors. Studies on the iLs of TPR have been conducted to identify the key G-protein-interfacing regions and residues. Mutational and protein truncation experiments have revealed that several regions contribute in varying degrees to G-protein binding/coupling. It was found that iL1, iL2, and iL3 as well as the intracellular C-terminal tail of TPR are critical for downstream signaling. 41,42 For example, it was demonstrated that mutation of Arg 60 within iL1 of TPR results in normal ligand binding but decreased second messenger production. 27 This suggests that Arg 60 is a key amino acid for Gq recognition. 30 Furthermore, it has been shown that Glu 129 and Arg 130 , which comprise part of the biologically important Glu-Arg-Tyr (ie, ERY) motif within iL2, are also important for Gq coupling. 43 Thus, given the limitations of mutational and truncation studies, at present controversy remains regarding the participation of the various intracellular TPR domains in G-protein coupling.
G-protein-Mediated Signaling Mechanisms of the Thromboxane A2 Receptor
Binding of a ligand to TPR results in a host of molecular responses that ultimately serve to elevate intracellular Ca2+ levels, an event that is central to platelet activation. 13 The first downstream event is stimulation of several G-protein isotypes. The Gq family (principally Gq in platelets but also G11, G15, and G16) 44 –46 activates PLC-β which mediates the conversion of phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). The second messenger IP3 serves to release intracellular calcium stores, and DAG activates protein kinase C 47,48 , both of which are critical in platelet aggregation. On the other hand, the G12 family (G12 principally but also G13 in platelets) 44,45 activates Rho guanine nucleotide exchange factor (RhoGEF), which regulates actin cytoskeletal remodeling, cytokinesis, cell motility, contraction, and myosin light chain kinase, 49 –51 leading to shape change in platelets. In terms of the G-protein-coupling profile, some debate 52 exists as to whether TPR couples with Gi, which inhibits AC activity. However, there is no evidence that TPR couples with Gs. Reports have also suggested a role for Gh, 53 which is a dimeric G-protein as opposed to the traditional trimeric model of G-proteins. Gh possesses transglutaminase activity that reportedly activates PLC-δ. 54 Regarding the heterotrimeric G-proteins, some have been found to associate with Gβγ subunits and are not simply quiescent regulatory elements. Rather, they have been shown to be involved in signal transduction processes such as activation of phosphatidylinositol 3-kinase (PI3 K), PLC-β2, and p44/42 mitogen-activated protein kinase (p44/42 MAPK)/extracellular signal-regulated kinase 1/2 (ERK1/2), 55,56 and so on. More recently, NMR techniques have been used to confirm studies that showed that both N- and C-terminal portions of the third intracellular loop (iL3), and the cytoplasmic tail of the receptors are involved in the G-protein activation and signal transduction. 27,28,29 Furthermore, the iL2, the membrane-proximal portions of the iLP3, and the N-terminal segment of the cytoplasmic tail contain amino acids that have been predicted to play a role in regulating the selectivity of G-protein and GPCR interactions. 57 –59
As an additional level of organization, GPCRs can form complexes with other GPCRs, thus modifying their presentation on the cell surface as well as their ligand specificity. 60 For example, in humans, TPR exists as two alternatively spliced variants, TPR-α 8 and TPR-β. 26 Since mice and rats only possess TPR-α, the sequence of which is similar to human TPR-α, it was initially thought that the α isoform is fundamental whereas β might have secondary functions. TPR-α and TPR-β have been shown to form homo- and hetero-oligomers, 61 a phenomenon that has several unexpected biological consequences. Thus, the TPR-α, 62 which does not normally undergo constitutive or agonist-induced internalization, can be internalized if TPR-α/TPR-β heterodimers form. This serves as a means of downregulating TPR signaling through the reduction in its surface presentation. Additionally, 63 TPR-α/TPR-β heterodimers were found to enhance isoprostane-mediated, but not (agonist)IBOP- or U46619-mediated, signaling, suggesting an alteration in the conformation of the agonist-binding pocket as a result of heterodimerization. It is interesting to note that when TPR-α and PGI2 receptors (IPR) 64 form heterodimers, it results in a “PGI2-like” response that was found to facilitate agonist/TPR-mediated cyclic adenosine monophosphate (cAMP) generation. Consistent with the previous observations of TPR-α/TPR-β heterodimers, IPR/TPR-α internalization has also been reported 63 as a consequence of dimer formation. On this basis, the notion of receptor dimerization should be considered a potential target for therapeutic TPR interventions.
Furthermore, recent findings suggest that when a G-protein is coupled with TPR, its downstream activity may be modified. 65 To this end, as a function of its structure, and specifically its iLs, the TPR protein can recognize only 1 G-protein at a time. However, receptor recycling has been shown to regulate what specific G-protein is docked at any given moment, and hence a “separate” G-protein may couple to the recycled receptors. The strength of a given TPR-G-protein association may vary with different G-proteins and may result in an increased probability for a specific pairing under certain conditions. Using TPR-α/Gα12 or TPR-α/Gα13 fusion proteins along with the nonhydrolyzable GTP analog GTPγS, U46619 is found to be a full agonist of TPR-α coupled with Gα12 and Gα13, in contrast to isoprostanes and pinane TXA2 (a stable analog of TXA2), which are “weaker” agonists for TPRα/Gα12. These findings indicate an alteration in binding specificity. However, this does not exclude the possibility of the formation/creation of alternative binding sites or even novel receptors, 12 which might contribute to differences in the ligand affinity for TPRs based on tissue expression (eg, platelets vs vascular smooth muscles). 29
Drug-Targeting of the Thromboxane A2 Receptor Pathway
Thromboxane A2 is an essential endogenous ligand for a host of biological processes and especially so in platelet functional responses. It is one of many by-products formed during COX-1-mediated metabolism of AA. 66 Platelets and other cellular tissues mediate TXA2 biological actions through TPR, 67 as previously mentioned. Both TXA2 and TPR play a role in a diverse set of physiological pathways and thus inappropriate or excessive activation of this modality can lead to a host of pathological conditions, 14 –16 such as myocardial infarction, thrombotic stroke, peripheral artery disease, embolism, and pulmonary dysfunction. In response to this prominent role in several disease states, extensive studies along several avenues elucidating the control of TXA2 activity has been investigated with varying degrees of success.
Cyclooxygenase/Thromboxane Synthase Inhibitors
Cyclooxygenase inhibitors such as nonsteroidal anti-inflammatory drugs (NSAIDs) and aspirin are currently the most extensively implemented method of TXA2 inhibition through their actions at the earliest level of biosynthesis. Cyclooxygenase inhibitors halt the production of TXA2 by blocking the conversion of AA to PGH2. At present, 3 COX isoenzymes are known to exist: (1) the stably expressed/constitutive COX-1, (2) the inducibly expressed COX-2, and (3) COX-3. Of note, COX-3, whose RNA is found in canines, has yet to be identified in humans. 68 It was found that COX-3 is a splice variant of COX-1, which retains intron 1 and has a frameshift mutation. Cyclooxygenase 3 is also known as COX-1b or COX-1 variant (COX-1v). 68 Platelets express COX-1, and nonselective COX inhibitors (acting on both COX-1 and COX-2) are effective at reducing TXA2 synthesis in platelets. Aspirin inhibits COX-1 through irreversible acetylation of a serine residue in the catalytic channel. 69,70 If used at higher concentrations, aspirin may also inhibit COX-2. 71,72 This activity translates into permanent suppression of TXA2 generation in platelets, as they lack a nucleus and therefore cannot regenerate COX-1. The presumed selectivity of low-dose aspirin is the underlying mechanism for its cardioprotective effects.
Unfortunately, in certain cases or patients, nonspecific COX inhibition by aspirin may result in the prevention of PGI2 synthesis, 73 a lipid that is necessary for the maintenance of the endothelium (ie, cytoprotective in nature). Consequently, while effective at inhibiting platelet function and reducing pain, chronic or high doses of aspirin can result in a host of deleterious side effects, including disruption of the gastrointestinal lining. In addition, the capacity of aspirin to inhibit the production of PGI2, is thought to be relevant to the understanding of why certain individuals in the population exhibit aspirin sensitivity as well as resistance. 21,73
Based on these considerations, research efforts have focused on modifying downstream effectors in TXA2 synthesis, as a preferable pharmacological approach. Thus, thromboxane synthase (TXS) inhibitors emerged as a viable means to prevent the conversion of PGH2 to TXA2. These drugs reduce TXA2 synthesis mainly in platelets, and have been found to improve TXA2-related pathophysiological conditions, such as thrombosis formation and thrombosis-related disorders. 74 In the last few decades, a plethora of TXS inhibitors were designed and tested for biological activity. For example, dazoxiben (UK37248), which is an imidazole derivative, was found to reduce the serum thromboxane B2 (TXB2) levels in human volunteers. Moreover, AA-induced platelet aggregation was completely inhibited by dazoxiben, whereas aggregation-elicited by adenosine diphosphate (ADP) was unaffected. 75 In terms of the ex vivo and in vivo effects, dazoxiben, reduced platelet activation in rats, and the markers for thrombin–fibrinogen interaction in the plasma. This in turn decelerated intravascular blood coagulation and increased thrombin tolerance. 76 Findings such as these show the importance of further studies of such inhibitors as potential new antiplatelets drugs.
Dazmagrel (UK38485), 77 another TXS inhibitor, has been shown to alleviate some of the harmful effects of ischemia in rats. 78 It was also found to reduce occlusions in the coronary artery of dogs. 77 As might be expected, this drug prevented TXA2 release that normally occurs during acute myocardial ischemia but did not suppress that of prostacyclin.
Pirmagrel (CGS13080) 79 specifically inhibited cell-free thromboxane synthetase (with an IC50 of 3 nmol/L); whereas submicromolar concentrations inhibited calcium ionophore-induced formation of TXB2, both in intact human platelets and in rats (when given at 1 mg/kg orally). Regarding its in vivo activity, data showed that pirmagrel is able to inhibit the synthesis of TXA2 by 80% to 75%, without any apparent effects on prostacyclin synthesis. 80 Therefore, this agent maintains the protective role of prostacyclin but still prevents uncontrolled platelet aggregation, which is a primary factor in the occlusion of blood vessels. 81
Another TXS inhibitor that was synthesized and examined is ozagrel (OKY046). 82 Intravenous injections (20 mg/kg) of ozagrel in beagles were shown to eliminate the cyclical reduction in coronary flow induced by vasoconstriction, which suggests a role for TXA2 in this process. CS-518 (RS5186) 83 is also a TXS inhibitor that was investigated in canine coronary occlusion and reperfusion models. It reduced the infarct size, the area of gross myocardial hemorrhage, and the intensity of neutrophil infiltration into the infarcted area. Isbogrel (CV4151) 84 delayed the initiation of hypertension in spontaneously hypertensive rats and reduced excretion of TXA2 (as TXB2) while having little effect on blood pressure and renal function. In isolated perfused kidneys of rats, isbogrel markedly inhibited both the AA-induced pressor action and the production of TXA2. Furegrelate (U63557A), a selective TXS inhibitor was found to be a potent inhibitor of TXA2 production in human platelets (in vitro) and rhesus monkey platelets ex vivo. 85 Similarly, it prevented the blockage of stenosed coronary arteries, in dogs. 85 Interestingly, U63557A does not appear to have a significant effect on hemostasis, 86 which would be an added benefit when compared to currently used, US Food and Drug Administration (FDA)–approved antiplatelet agents. It is noteworthy that TXS inhibitors have been found to indirectly increase PGI2 levels by making more AA available, thus further inhibiting platelet activation. Unfortunately, this class of drugs failed in clinical settings. The underlying reason for this failure is the accumulation of PGH2, which exerts agonistic activity on TPR, thereby increasing platelet activity. 87
Thromboxane A2 Receptor Antagonists
Given the failure of the TXS inhibitor class of drugs, attention turned to the TPR itself as a possible therapeutic target. This class of drugs would antagonize the effects of TXA2 on TPR while preserving endothelial production of PGI2. Another advantage for the direct antagonism of TPR over TXA2 synthesis inhibition is that such drugs would not only block the effects of TXA2, but also those of known ligands such as endoperoxides and isoprostanes. 88,89 Taken together, it is clear that to limit undesirable side effects while maximizing drug efficacy, direct targeting/antagonizing of TXA2 binding to TPR would be a much preferred route of intervention.
Unfortunately, following efficacy and toxicity issues, only modest success has been reported in developing pharmaceutics along this avenue of research. The majority of discovered agents have been discontinued at phase I/II clinical trials. Thus, TPR antagonists remain as pharmacological research tools for investigating receptor-mediated signaling, biology, and activating events.
To this end, SQ29,548, a highly selective TPR antagonist, is the gold standard with regard to TPR antagonists, 24 and has greatly contributed to the elucidation of TPR-mediated signaling events. It has the ability to inhibit U46619-induced aggregation of human platelets, and contraction of rat and guinea pig tracheal, arterial, and venous smooth muscles. It also inhibited the contraction of rat vascular smooth muscle induced by 8-iso-PGF2 α. Domitroban (S-1452/S-145) 90 possesses TPR-dependent pharmacological effects that involve animal models of disease states such as asthma, rhinitis, and brain ischemia. Additionally, it was found to be very selective with high binding affinity for TPR, and hence it was utilized to purify the receptor protein using affinity chromatography. 91
Although the majority of TPR antagonists fail to make it into clinical practice, development of these drugs for treatment of various TPR-dependent disorders has continued. Thus, daltroban (BM13505) 92 has been found to antagonize the binding of [ 3 H]SQ29,548 to human platelets, 93 which translated to attenuation of U46619-induced aggregation of human platelets. Ifetroban (BMS180,291) 94 is a very potent, long-acting antagonist, which was found to exhibit improved metabolic stability and oral bioavailability. Additionally, it appeared to be highly selective and display high affinity for human platelet TPR, which was documented in competition-binding experiments with [ 3 H]SQ29,548. Ramatoroban (BAY-u3405) 95 is another selective antagonist, which was found to exhibit in vitro antagonistic effects on U46619-induced constriction of human and rodent smooth muscle cells, as well as bronchoconstriction, in guinea pigs, in vivo. 95 In addition, this drug is currently marketed, in Japan, for use in allergic rhinitis, due to its ability to attenuate PGD2-induced bronchial hyperresponsiveness (in humans). Seratrodast (AA-2414) 96 is yet another orally active TPR antagonist that has been used clinically for the treatment of asthma in Japan.
Sulotroban (BM13177) 25 is a PGH2 receptor/TPR antagonist that was not modeled on the chemistry of TXA2 (ie, structurally dissimilar), but it was shown to selectively block TPR on platelets and vascular smooth muscle 25,97 –99 . Terutroban (S18886) 100,101 is an oral, TPR-specific antagonist that was found to inhibit thrombus formation, possess antiproliferative and antiatherogenic activities 102 –104 in animal models. In coronary patients receiving aspirin, terutroban resulted in improved endothelial dysfunction. 105 This finding suggested that combination of aspirin and a TPR antagonist may result in added benefit and better-therapeutic outcomes. Vapiprost (GR32191) 106 showed potential as a TPR antagonist as it antagonized both shape change in platelets and aggregation in response to U46619. Z-335, 93 is another orally active TPR antagonist that inhibited the binding of [ 3 H]SQ29,548 to human platelets, and U46619-induced human platelet aggregation in vitro. 93 Interestingly, this compound did not prolong bleeding in animals, which would allow for addressing the risk of thrombosis without bleeding complications.
Clearly, a great deal of work has gone into adapting experimentally derived TPR antagonists into clinical interventions. Unfortunately, the vast majority of these compounds fail to pass clinical trials or toxicity screening as mentioned previously. Two somewhat unorthodox alternatives to bypass these obstacles include isolation of naturally occurring compounds or to repurpose currently FDA-approved compounds for use as TPR antagonists. A small, but growing body of agents has been isolated from plants that appear to possess the potential for antagonizing TPR. These alternative approaches may be somewhat fitting, given the most commonly used inhibitor of TPR signaling/activity being aspirin, which was originally derived from a natural source, the willow bark. Genistein 107 is a naturally occurring isoflavone found in soy and which suppressed platelet aggregation in vitro, and in an in vivo mouse model it was reported to prevent thrombogenesis. In fact, the flavonoids have multiple members that have been reported as TPR antagonists. 108 Similarly, apigenin, luteolin from celery, quercetin from capers, and kaempferol from tea have also been reported to show antagonistic activities toward TPR. 108 The capacity of flavonoids to interact with TPRs in platelet-rich plasma (PRP) was initially explored using radioligand ([ 3 H]-SQ29,548)-binding assays. Furthermore, in perfusion studies, it was documented that apigenin, genistein, and catechin significantly diminished thrombus formation.
In addition to flavonoids, piperlongumine, 109 a constituent of long pepper, showed antagonistic activity toward TPR. In this connection, Piper longum L. has been used as a crude drug for the treatment of poor peripheral blood circulation in Asia. In terms of its biological activity, piperlongumine inhibited U46619-triggered platelet aggregation, in a dose-dependent fashion, whereas it only slightly inhibited the thrombin-induced response. Piperlongumine also inhibited U46619-induced phosphatidylinositol hydrolysis and the binding of [ 3 H]SQ29,548 to TPR, in a similar concentration-dependent manner. Finally, and interestingly, reports 110 have indicated that phenol red, which is used extensively in cell culture models, has the capacity to antagonize TPRs, even at concentrations at which it is used as a pH indicator. Specifically, phenol red 110 relaxed canine lingual arteries that were contracted with U46619 and inhibited U46619-induced aggregation in human platelets, in a manner similar to the TPR antagonist ONO-3708. Thus, careful consideration should be given when designing experiments investigating TPR signaling or biology to exclude phenol red as a pH indicator.
Combinatorial Drugs
A relatively recent development in the efforts to target the thromboxane pathway is the discovery of drugs that serve multiple purposes; for example, the inhibition of TXA2 synthesis and the antagonism of TPR. This approach would theoretically result in a “superior” drug that can probably be given at lower doses, thereby reducing potential toxicity and adverse effects. To this end, picotamide (G137), 97 a derivative of methoxy-isophtalic acid, has been reported to antagonize TPRs as well as inhibit TXS. It was found to suppress the aggregation of human platelets induced by AA, U46619, low-dose collagen, as well as authentic TXA2, without affecting the aggregation induced by A23187 or ADP. A separate drug of this class is known as ridogrel (R68070) 111 and is used with streptokinase as an adjunctive therapy to reduce the formation and size of blood clots. A comparison between aspirin and ridogrel as an adjunct to thrombolysis in patients with acute myocardial infarction (MI) demonstrated that while ridogrel is not superior to aspirin in enhancing the fibrinolytic efficacy of streptokinase, it may in fact be more effective in preventing new ischemic events. Unfortunately, clinical experience with this drug is still relatively limited. Terbogrel, 112 E-6-(3-(2-cyano-3-tert-butyl-guanidino)phenyl)-6-(3-pyridyl)hex-5-enoic acid, inhibited TXS and, displaced radiolabeled [ 3 H]SQ29,548 in human platelets. 112 Additionally, terbogrel was found to inhibit collagen-induced platelet aggregation using both human PRP and whole blood. These inhibitory effects were shown to translate into a potent antithrombotic effect in vivo, as demonstrated in studies using a model of arterial thrombosis in rabbits. Diclofenac (Voltaren) 113 is a highly potent, nonspecific COX inhibitor that was reported to competitively inhibit TPR. In addition, it was found that this antagonistic activity was shared with its derivative, the highly selective COX-2 inhibitor known as lumiracoxib. Interestingly, diclofenac has been linked to other modes of action. Thus, there is evidence suggesting that it may inhibit the lipoxygenase enzyme pathway (production of leukotrienes [LTs]), PLC-A2, voltage-dependent sodium channels as well as other ion channels. These findings remain to be confirmed and may be the focus of further investigations.
Torasemide (Demadex) 114 is a pyridine-sulfonylurea type loop diuretic typically used in the management of edema associated with congestive heart failure. At low doses, it is used for the management of hypertension. It was found to inhibit TXA2-mediated vasoconstriction; and based on this finding, several derivatives have since been developed. One such derivative is BM-520 (N-tert-butyl-N′-[2-(3′-methylphenylamino)-5-nitrobenzenesulfonyl]urea), which was found 115 to (1) effectively displace radiolabeled [ 3 H]SQ29,548; (2) inhibit AA- and U46619-induced human platelet aggregation; and (3) inhibit U46619-mediated rat aortic smooth muscle contractions, though not to the degree of SQ29,548. 115 Subsequently, this effect was confirmed through in vivo studies, where a single dose of BM-520 given to guinea pigs revealed that their platelets required significantly greater concentrations of AA and U46619 (compared to control-treated guinea pigs) for activation.
Another antagonist, BM-531 114 (N-tert-butyl-N′-[(2-cyclohexylamino-5-nitrobenzene)sulfonyl]urea) exhibited an affinity for human platelet TPRs that is higher than SQ29,548. Consequently, BM-531 was characterized by a potent inhibition of platelet aggregation in response to AA, U46619, and collagen. It was also found to inhibit the second wave of ADP-induced aggregation. Moreover, BM-531 can totally halt production of TXB2 by human platelets that are activated by AA. Of note, BM-531 is devoid of the diuretic property of torasemide and thus can be regarded a promising antiplatelet agent. Another derivative is BM-567 116 (N-pentyl-N′-[2-cyclohexylamino-5-nitrobenzene)sulfonyl]urea), which showed a higher affinity for TPR than BM-531 (IC50: 7.8 ± 0.7 nmol/L) and sulotroban. Similar to BM-351, it also prevented platelet aggregation induced by AA, U46619, and collagen, inhibited the second wave of ADP, and halted TXB2 production in human platelets. 116 Another torasemide derivative is BM-573, 117 which possesses a high affinity for human platelet TPR in comparison with SQ29,548. Specifically, it was found to be a potent inhibitor of human platelet aggregation and the production of TXB2 induced by AA and U46619. 117 As an alternative combinatorial drug, TRA-418 118 was developed as a novel TPR antagonist with an additional IPR agonistic activity. It was found that TRA-418 inhibited platelet aggregation induced by the TPR agonist U46619 and by AA at concentrations lower than those required for inhibition of ADP-induced aggregations. Furthermore, TRA-418 inhibited platelet aggregation induced by ADP alone and that induced by ADP in the presence of the TPR antagonist, SQ-29,548. 118 YM158 119 is another type of dual-acting drugs antagonizing TPR and LTD4 receptors, which was found, using a guinea pig model, to be effective in relaxing bronchial smooth muscle. 120
It has been suggested that in addition to their principal activity, angiotensin II receptor blockers (ARBs) also behave as TPR/PGH2 receptor antagonists. In particular, losartan 121,122 and irbesartan 123 can selectively inhibit the in vitro contraction elicited by the TXA2 analogue U46619 in many animal- and human isolated-vascular preparations. Losartan was found to significantly inhibit human platelet activation in vitro (i.e., reduced platelet activation due to ADP and thrombin in an angiotensin II receptor–independent manner). Ex vivo data (unpublished) using rodent platelets revealed that losartan selectively inhibits aggregation mediated by TPRs. Thus, the exact mechanism by which these molecules exert antiplatelet effects appears to be somewhat controversial but might be explained by differing experimental conditions. Specifically, earlier studies were done in the absence of COX-1 inhibition, and inhibiting the propagation of signal due to secreted TXA2. On this basis, it remains that antagonizing TPR is in fact the predicted mechanism for this phenomenon (unpublished data). Further evidence in support of this modality of inhibition was provided in another study where unlabeled losartan competitively displaced [ 3 H]U46619 from human platelets. 102 Irbesartan has been less intensely investigated, but it also appears to specifically inhibit TPR as it abrogated platelet activation as well as contraction of isolated human saphenous vein sections, elicited by U46619. 121
Fenofibrate, 124,125 a fibric acid derivative, has a host of effects that together serve to lower lipid levels. Interestingly, it has also been associated with antithrombotic effects, that is a marked increase in time to occlusion in a ferric chloride artery injury model in rats administered fenofibrate orally for a week. As might be expected, this regimen resulted in a decrease in platelet aggregation. The mechanism for this antiplatelet activity appears to be a result of a significant decrease in platelet calcium mobilization, blockade of TPR, and inhibition of COX-1 activity. KP-496 126 , (2-[N-[4-(4-chlorobenzenesulfonylamino)butyl]-N-[3-[(4-isopropylthiazol-2-yl)methoxy]benzyl]sulfamoyl]benzoic acid), is a dual antagonist for the cysteinyl leukotriene receptor 1 and for TPR. It was rationally designed based on the evidence that cysteinylleukotrienes and TXA2 contribute to the recruitment of eosinophils into the lung, in asthmatics. These effects are thought to be the underlying mechanism for some of its anti-inflammatory properties.
The Drug Rediscovery Approach
In addition to the aforementioned compounds, drugs that have already been approved for clinical use may hold the potential for use as TPR antagonists despite being often prescribed for completely different and unrelated indications. It might be rather surprising and peculiar that while the classical drug discovery is yet to produce a TPR antagonist into clinical practice, some of the currently FDA-approved drugs (as also previously indicated) have been found to interact with/inhibit TPRs, in both selective and nonselective manners. One such drug is glybenclamide, which is a sulfonylurea, antidiabetic drug which inhibits ATP-sensitive potassium channels in pancreatic beta cells. 127 It was observed that its chemical structure contains somewhat identical pharmacophores and orientation/presentation as those of the classical TPR SQ29,548, suggesting the potential of being repurposed for use as a TPR antagonist. In support of this notion, glybenclamide was found to antagonize responses to U46619 in rat aorta and mesenteric artery, yet failed to inhibit contractions in guinea pig vessels. 128 Moreover, the combination of glybenclamide and GR32191 (see above) caused a rightward shift in the concentration–effect curve of U46619 in rat aorta, indicating that these 2 drugs produce additive effects. Finally, and most recently, published work has confirmed in vitro that glybenclamide exerts TPR-specific inhibition of platelet aggregation, as it abrogated the response to AA and U46619, while having no effect on aggregation mediated by ADP and TRAP4. 129 These findings were later confirmed under ex vivo platelet aggregation experimental settings, and using an in vivo mouse model of thrombosis. 130
Conclusion
Clearly, elucidating the structure, function, signaling mechanisms, and potential interventions for TPR has been the focus of a great deal of work over the course of decades. Despite this body of knowledge, there is a dearth of clinical options to serve as antiplatelet agents focusing on this specific axis of activation, particularly at the point of ligand binding to TPR. As this is the option with the lowest potential for side effects as observed with aspirin, or with potentially inefficient inhibition as with TXS inhibitors, there is great interest in developing direct TPR antagonists. Unfortunately, most of the work to date has run into difficulties at the stage of clinical trials due to efficacy and toxicity issues.
On the other hand, human and animal studies clearly show that certain FDA-approved drugs have the capacity to selectively block TPR signaling/function, which opens the door for possibly repurposing them, or designing a more potent structural derivative. Finally, we hope that the present overview of the thromboxane receptor, briefly detailing studies investigating ligand binding and G-protein coupling, as well as the work to date on TXA2/TPR interventions would provide a knowledge base by which development of novel TPR antagonists can be furthered.
Footnotes
Acknowledgment
The authors would like to thank Dr Wallace J. Murray for proofreading and editing the manuscript.
The first two authors contributed equally to this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by funds provided by The Regents of the University of California, Tobacco-Related Disease Research Program, Grant Number 19KT-0030 (to FTK). The opinions, findings and conclusions herein are those of the author and not necessarily represent those The Regents of the University of California, or any of its programs. This work was also supported by Intramural Funding from the College of Pharmacy at Western University of Health Sciences (to FTK).