
Abstract
The purpose of this article is to review the basis of arrhythmogenesis, the functional and clinical role of the late Na current, and its therapeutic inhibition. Under pathological conditions such as ischemia and heart failure this current is abnormally enhanced and influences cellular electrophysiology as a proarrhythmic substrate in myocardial pathology. Ranolazine the only approved late Na current blocker has been demonstrated to produce antiarrhythmic effects in the atria and the ventricle. We summarize recent experimental and clinical studies of ranolazine and other experimental late Na current blockers and discuss the significance of the available data.
Antiarrhythmic Therapy
Cardiac arrhythmias are a leading cause of death in the Western world, particularly ventricular tachyarrhythmias, leading to ventricular fibrillation and sudden cardiac death. Many patients with ventricular tachyarrhythmias have structural heart disease, which is often associated with abnormalities of impulse generation and electrical conduction. Severe ventricular arrhythmias can occur in participants with ischemic and nonischemic, mainly dilated, cardiomyopathies, congenital heart disease, and various genetically and acquired abnormalities of the myocardium. Atrial fibrillation (AF), on the other hand, is the most common sustained heart rhythm disorder and is often considered a disease of aging, thereby causing a serious public health problem. Epidemiological data clearly show that incidence and prevalence of AF will dramatically increase in the future. Complications due to AF include, among others, embolic stroke and heart failure, thereby further increasing morbidity and mortality. With an ever-aging population, especially in developed countries, finding effective, reliable, and safe treatments for atrial and ventricular arrhythmias, aside from its primary prevention, is of utmost importance.
Amiodarone is used to treat the majority of patients with significant structural heart disease who require antiarrhythmic drug treatment. Although effective against AF and ventricular arrhythmias, it has been associated with serious extracardiac side effects in a variety of tissues. 1 As a consequence, there has been much research in the quest to identify safer treatments for AF. Recently, evidence from clinical trials has highlighted the potential antiarrhythmic efficacy of ranolazine, a sodium channel blocker with rapid unbinding kinetics. 2 –5 Furthermore, ranolazine displays a favorable safety profile even in patients with structural heart disease and has already been approved for the treatment of chronic angina. 6,7 This review focuses on the role of the late sodium current, INa,late, in arrhythmogenesis and the current evidence suggesting a potential antiarrhythmic role for ranolazine and its mechanisms of action.
Characteristics of INa,late
The cardiac action potential (AP) is initiated by the opening of sarcolemmal voltage-gated Na+ channels, mainly Nav1.5, leading to the influx of Na+ ions down an electrochemical gradient. The majority of these channels are only transiently active, rapidly inactivating with depolarizing voltage, and they carry the peak Na+ current (INa,peak). The density of INa,peak is greater in atrial compared to ventricular myocytes. However, owing to the fact that this sodium channel has a more negative steady state inactivation voltage in the atria 8 and the atrial resting membrane potential (RMP) is less negative compared to ventricle, the actual amount of available current is similar in atria versus ventricle. 9 A small fraction of these cardiac Na+ channels has alternative gating 10,11 and carries the late Na+ current (INa,late). Sodium channels producing this additional current are thought to remain active over a longer period (inactivating more slowly) or even to reopen 12 and can be found in the atria and ventricles. Although the amplitude of INa,late is much smaller than that of INa,peak, its persistent nature during the AP means that it may contribute significantly to Na+ loading in the cell and AP duration (APD) prolongation. 12 –17 Indeed, congenital long-QT syndrome type 3 (LQT3) is caused by a gain-of-function mutation of the gene SCN5a that encodes the sodium channel Nav1.5. 18
INa,late and Arrhythmogenesis
Under several pathological conditions, INa,late is increased and evidence for this has been found in human and also in animal models of cardiac disease, including hypoxia, ischemia, hypertrophy, myocardial infarction, heart failure, and AF. 16,19 –26 Intracellular Na+ overload plays an important role in heart failure 27,28 and can alter both systolic and diastolic function via effects on intracellular Ca2+. 17,29 –32 Increased INa,late is also thought to lead to arrhythmogenic activity, 33 and this shall be focused on in more detail.
The mechanisms of cardiac arrhythmia can be separated broadly into 2 groups: abnormal impulse formation and abnormal conduction. Abnormal impulse formation includes abnormal automaticity, such as spontaneous diastolic depolarizations (DDs), which can lead to tachyarrhythmias. Also under the umbrella term of abnormal impulse formation is triggered activity. Triggered activity is caused by instability of the cell membrane potential and formation of afterdepolarizations either during the AP (early afterdepolarization [EAD] phase 2 or phase 3, respectively, depending on the time during the AP at which it occurs) or after the AP repolarization (delayed afterdepolarization [DAD]), as subsequently discussed.
INa,late has been shown to contribute to abnormal automaticity, and its inhibition by ranolazine can suppress DD and spontaneous APs. 34 Sodium ion influx via INa,late also leads to APD prolongation. 12,13,16,35 When sufficient APD prolongation occurs, inward currents are able to recover from inactivation, become reactivated, and allow charge to enter the cell. If current entering the cell exceeds repolarizing K+ currents, an EAD can occur. Ca2+ entering the myocyte during the AP plateau can lead to an inward current on forward mode sodium–calcium exchange (NCX), which can prolong the AP further 36 and, together with enhanced INa,late, may contribute to the EAD upstroke. 33 In addition, Ca2+ entering the cell activates Ca2+/calmodulin-dependent protein kinase II (CaMKII) which, in turn, increases INa,late via phosphorylation of the sodium channel, as discussed later. The importance of INa,late for APD prolongation and the incidence of EADs are supported by the finding that inhibition of INa,late by ranolazine reduces APD and EAD occurrence in various settings where INa,late is enhanced. 22,37 –39 Early afterdepolarizations, depending on the membrane potential at which they occur, can trigger APs and ectopic activity that, if propagated, appears as extra beats on the electrocardiogram. 40 Hearts from mutant mice, delta KPQ SCN5a, which express a “gain of function” Na+ channel, mimicking human LQT3 syndrome, display EADs and ventricular tachycardia, highlighting the contribution of Na+ influx to this arrhythmogenic mechanism. 41 Furthermore, triggered activity, caused by EADs in the pulmonary vein, is believed to play a significant role in the induction of AF. 42
Increased INa,late is thought to indirectly cause DADs; 33 enhanced INa,late causes intracellular sodium overload, which leads to calcium overload, via reverse mode NCX current. 17,29,43 –45 The resulting calcium overload is thought to trigger intracellular Ca2+ release from the sarco–endoplasmic reticulum (SR), leading to cytosolic Ca2+ oscillations, automaticity, and triggered activity. 16,35,46 –48 Calcium overload and oscillatory activity are able to activate forward mode NCX. 49 This carries an inward positive current, also known as the transient inward current due to NCX stoichiometry (1Ca2+:3Na+), thereby bringing about a DAD. 50 –52 If a DAD reaches threshold for an AP to occur, triggered activity and extrasystolic activity follow. 51,53 Pharmacological enhancement of INa,late has been shown to induce DADs and triggered activity. 48,53 Furthermore, inhibition of INa,late by ranolazine reduces the incidence of DADs and prevents triggered activity. 16,48,53 –55 Delayed afterdepolarizations have been observed in several types of cardiac tissue, including both atrial and ventricular myocytes, where they can lead to triggered activity and tachyarrhythmias. 56
The second mechanism of arrhythmia in the heart is abnormal conduction and includes conduction block and reentry. Reentry is a common cause of arrhythmia and can be observed and experimentally induced in various locations, including the atria and ventricles. 57,58 Reentry is brought about by spatiotemporal heterogeneity of repolarization and conduction velocity (CV) and unidirectional block, which acts as a substrate for new circuits of conduction to form in the myocardium. Reentrant activity can also result from EADs upon the development of electrical heterogeneity 40,59 and is associated with the development of torsades de pointes (TdP). 60 INa,late contributes to APD, and considering its higher density in Purkinje fibers and midmyocardial (M) cells versus epicardium, it is an important determinant of repolarization heterogeneity. Pharmacological enhancement and inhibition of INa,late have been shown to increase and decrease transmural dispersion of repolarization (TDR), respectively, as reviewed by Antzelevitch and Belardinelli. 61 Experiments to mimic LQT3 in canine ventricular wedge preparations have shown that enhancement of INa,late by the sea anemone toxin (ATX-II) causes a preferential APD prolongation in M cells compared to other myocyte types, thereby increasing TDR. Block of INa,late has been shown to completely suppress TdP in various models. 62,63 Furthermore, pharmacological inhibition of INa,late reduces APD variability in myocytes from failing human hearts (where INa,late density is increased) 64 and has also been observed to suppress reentry in a rat model of ventricular fibrillation. 65
Abnormalities in sodium and calcium handling play a crucial role in various cardiac pathologies. In light of the finding that INa,late is increased in many of these conditions and that its inhibition suppresses a variety of arrhythmias, specific block of this current has been suggested as a potential therapy in the treatment of cardiac pathologies, in particular as part of a multichannel blocking approach. 44,66 –72
INa,late and CaMKII
INa,late is thought to be modulated in several ways, including by toxins (for example ATX-II), hypoxia, ischemia, reactive oxygen species, the expression of different channel subunits, and also CaMKII. 20,21,55,73 –79 Calmodulin-dependent protein kinase II is a signaling molecule, and the predominant isoform in the heart is CaMKIIδc. It has been shown to be a key protein in the regulation of various aspects of cardiac excitation–contraction coupling. 80 Calmodulin-dependent protein kinase II is activated by Ca2+-calmodulin complex (Ca2+/CaM), and activity is further enhanced by autophosphorylation. Expression and activity of CaMKII are upregulated in heart failure and hypertrophic cardiomyopathy, as shown in animal models and in human disease. 22,81 –86 Calmodulin-dependent protein kinase II expression is also increased in AF. 87,88 Overexpression of CaMKIIδc has been shown to cause arrhythmias, hypertrophy, and diastolic dysfunction, implying that CaMKII upregulation may play a significant role in the manifestation of cardiac pathologies and heart failure. 55,77,82,89 –91
There is extensive evidence that CaMKII phosphorylates the ryanodine receptor (RyR) at Ser2815, altering its open probability. 92 In particular, increased SR Ca2+ leak via phosphorylated RyR is believed to be associated with heart failure, 81,93 –97 and CaMKII has been shown to mediate this increase in Ca2+ leak. 81,85,86,98,99 Furthermore, in a mouse model of CaMKIIδ overexpression, inhibition of CaMKII was shown to suppress isoprenaline-induced increases in diastolic leak and arrhythmias. 91 Ryanodine receptor instability and increased SR Ca2+ leak are also considered to play an important role in AF 50,87,100 with CaMKII, again implicated as a significant mediator. 35,87
It is becoming increasingly clear that upregulated INa,late, intracellular Na+ overload, and increased activity of CaMKII are not acting alone, but rather in concert in cardiac disease. Acute overexpression of CaMKIIδc has been shown to facilitate INa,late by altering channel gating. 77 Furthermore, there is evidence that CaMKII coimmunoprecipitates with and phosphorylates Nav1.5; various phosphorylation sites on Nav1.5 have been reported for channel regulation by CaMKII. 76,101,102 The changes thought to occur in sodium channel gating upon phosphorylation by CaMKII include a shift to intermediate gating and enhanced INa,late with slowed recovery of INa from inactivation. 77 Indeed in the case that CaMKII is upregulated in heart failure and AF, one could speculate that these CaMKII-dependent shifts in sodium channel gating could prove importance in relation to ranolazine application, as there is evidence that ranolazine preferentially blocks the sodium channel in the inactivated state. 103 Conversely, however, there is also evidence for active state channel block by ranolazine. 104 Ranolazine has been shown to protect against CaMKII-induced premature arrhythmogenic contractions and impaired diastolic relaxation in a mouse model of CaMKIIδc overexpression. 55 Likewise, ranolazine has been observed in vitro to have antiarrhythmic effects and to protect against diastolic dysfunction in human myocardium from AF, with CaMKII implicated as a potential modulator of INa,late. 19
Evidence is also emerging that CaMKII plays a central role, downstream of INa,late and cytosolic Na+ overload, in the initiation of proarrhythmic SR Ca2+ leak. It has been shown that INa,late is associated with CaMKII activation and that the ensuing effects can be reversed by the inhibition of INa,late or CaMKII. 35,48,105 The associations between INa,late and CaMKII are thought to enable a vicious cycle to occur upon intracellular sodium and calcium overload, leading to chronic CaMKII activity 96 ; CaMKII increases both SR Ca2+ leak and INa,late (leading to further Ca2+ overload via reverse mode NCX), which positively feeds back on CaMKII activity, as illustrated in Figures 1 and 2. Considering the proarrhythmic risks associated with CaMKII overactivity, CaMKII inhibition has been suggested as an attractive therapeutic strategy in the treatment of arrhythmias and heart failure. 106 This approach is, however, hindered by the fact that CaMKII is found at high levels in other tissues and CaMKII inhibitors are not currently tissue-specific.
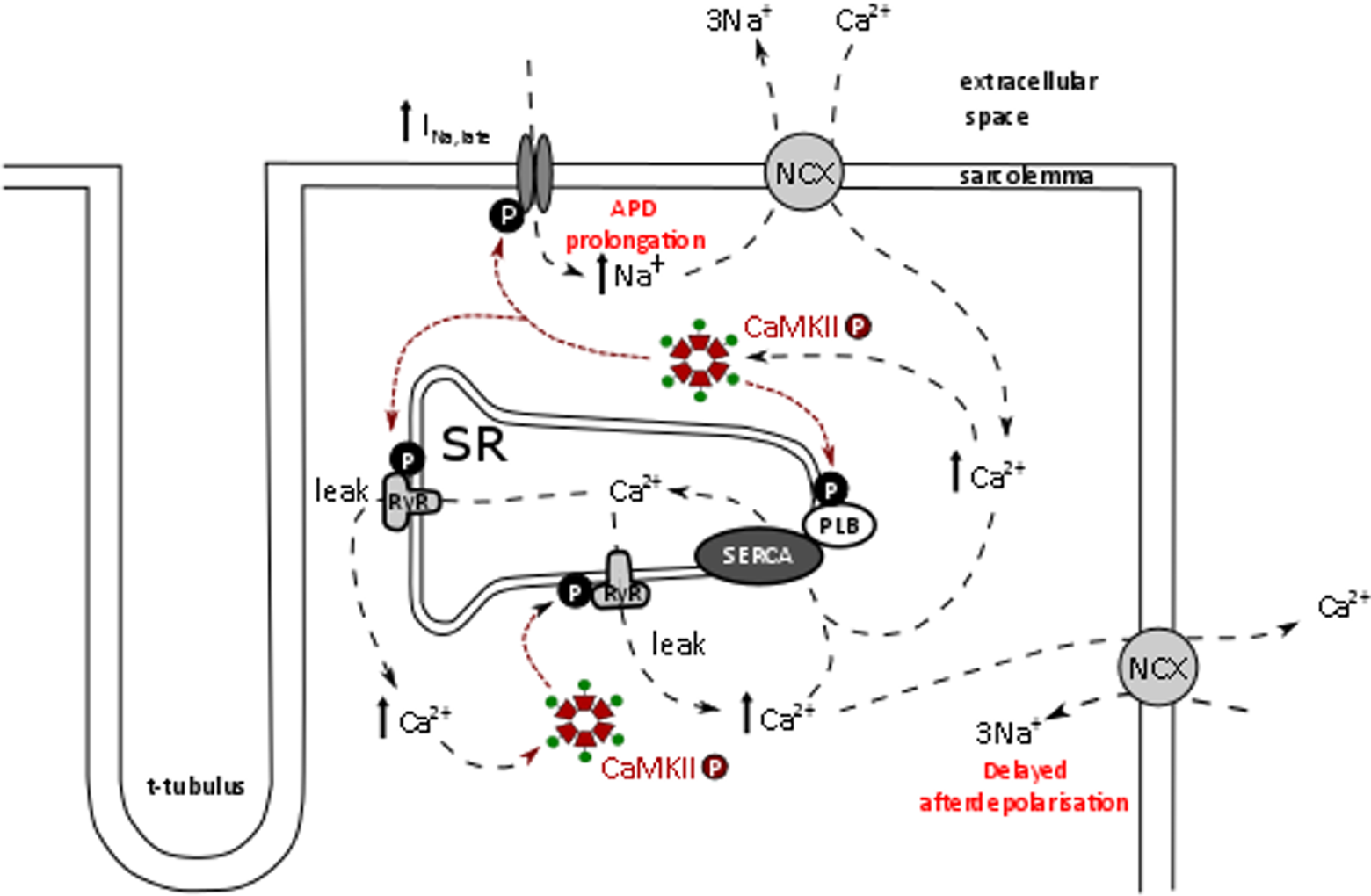
Sodium and calcium handling in the cardiac myocyte and activation of calmodulin-dependent protein kinase II (CaMKII) upon an increase in the late Na+ current (INa,late). Increased INa,late leads to sodium–calcium overload (reverse mode sodium–calcium exchange [NCX]). Ca2+ activates CaMKII that enhances INa,late further and also increases sarco–endoplasmic reticulum (SR) Ca2+ leak, which can lead to triggered activity via forward mode NCX. A vicious cycle is enabled, leading to chronic CaMKII activity.

The arrhythmogenic effects of increased sodium influx via late Na+ current (INa,late) in the cardiac myocyte. Increased INa,late leads to intracellular sodium overload that causes Ca2+ overload via reverse mode sodium–calcium exchange (NCX). Increased intracellular (Ca2+) causes increased calmodulin-dependent protein kinase II (CaMKII) activity and sarco–endoplasmic reticulum (SR) Ca2+ leak, which can lead to triggered activity and delayed afterdepolarizations (DADs), by activating a transient inward current (ITI) on the NCX. Increased INa,late also leads to AP duration (APD) prolongation that can give rise to early afterdepolarizations (EADs) due to recovery and reactivation of inward currents during the action potential (AP) plateau.
Ranolazine—A Potential Antiarrhythmic Agent?
The aim in the pharmacological treatment of AF is to terminate it and prevent its recurrence by reducing automaticity, CV, and reentry. Inhibition of the peak sodium current affects both excitation threshold and AP upstroke velocity (Vmax). Premature excitation can also be prevented by prolonging the effective refractory period past the APD, known as postrepolarization refractoriness (PRR). 107 Despite the efficacy of amiodarone in suppressing AF in patients with structural heart disease, it can cause severe organ toxicity and has a limited proarrhythmic risk at the ventricular level. This has prompted the search for other atria-targeted compounds.
Ranolazine is a unique drug used for the treatment of angina by acting as an anti-ischemic agent. 37,108 Originally, it was thought that the mechanism underlying this action of ranolazine may be the inhibition of fatty acid oxidation. 109 However, it has since become clear that a main site of action responsible for anti-ischemic and antianginal effects of ranolazine is the inhibition of INa,late. There are some studies that have shown β-adrenoceptor blocking properties of ranolazine, 110 including a very recent clinical study reporting a weak but significant effect of high-dose ranolazine (1000 mg) on heart rate in patients with angina in the absence of significant coronary heart disease. 111 Others have found that ranolazine at plasma concentrations lower than 10 μmol/L has only minimal antiadrenergic effects. 112 Therefore, the antiarrhythmic potential of ranolazine via β-adrenoceptor blockade remains a controversial issue and is likely highly dependent on dosage.
Ranolazine, like amiodarone, binds with rapid unbinding kinetics to the sodium channel, and as mentioned, there is evidence for both inactive state 103 and active state block. 104 The safety of ranolazine was demonstrated, among other studies, in the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) trial, 113 where it was also observed that ranolazine has potent antiarrhythmic capacities. 3 In this trial, ranolazine potently reduced nonsustained ventricular arrhythmias and also significantly suppressed supraventricular tachycardias. 3 Moreover, patients treated with ranolazine showed a tendency toward reduced new-onset AF (P = .08), although the incidence of AF was relatively low in this trial. 4
All sodium channel blockers are able to inhibit both INa,late and INa,peak. There is evidence that in the failing ventricle, inhibition of INa,peak by ranolazine is much less potent than that of INa,late, with IC50 values of 6.5 μmol/L (INa,late) and 244 μmol/L (INa,peak), 114 although this remains controversial as some reports show inhibition of INa,peak also in the ventricle by ranolazine. 115 In contrast, ranolazine is a powerful inhibitor of atrial INa,peak. 8,114 This “atrial-selective block” of INa,peak is thought to contribute to the effective antiarrhythmic action of ranolazine in the atria, without causing deleterious effects in the ventricle, such as reduced excitability. Ranolazine, like amiodarone, also blocks ‘rapid’ delayed rectifier current (IKr) in both the atria and ventricles, where the half maximal inhibitory concentration (IC50) is approximately 11.5 μmol/L. 114 The combination of sodium and potassium channel block by ranolazine has been shown to be effective against ventricular arrhythmias, as first shown in a guinea pig model of long-QT syndrome. 38 Furthermore, the anti-AF potential of ranolazine has been shown in animal 116 and human 2 –4,117 studies. Of note, the human study by Miles et al was conducted postoperatively and therefore concerns a form of AF with alternative characteristics, which could be considered a study limitation. Evidence that ranolazine is protective against both AF and ventricular arrhythmias has been reviewed by Verrier et al 118 and has led to the idea that ranolazine could be a more desirable option in the treatment of AF compared with other agents that are proarrhythmic in the ventricle, in particular, in patients with structural heart disease. Atrial fibrillation and heart failure can often occur together, and importantly, Burashnikov et al showed in canine wedge preparations that ranolazine can effectively suppress AF in the setting of tachypacing-induced heart failure. 119 In this study, parameters dependent on INa,peak, such as Vmax, CV, and PRR, were observed to be altered by ranolazine in the atria but not in ventricle. The effect of ranolazine was greater in the atria from failing than nonfailing hearts. Furthermore, ranolazine had no proarrhythmic effect on ventricular tissue. This combination of arrhythmia suppression in the atria, while excitability is maintained in the ventricles, represents a promising pharmacological approach for the handling of AF. The next section discusses in more detail the atrial and ventricular antiarrhythmic actions of ranolazine and the evidence to support ranolazine as a promising new option in the treatment of AF.
As described, fractional block of sodium channels by ranolazine is believed to be greater in the atria compared to the ventricles. This is thought to be due to electrophysiological differences between atrial and ventricular tissue 8 with the result that, in the atria, a higher proportion of sodium channels are in the inactivated state at a given resting or takeoff potential and there is evidence that ranolazine predominantly binds to this channel configuration. 103 In comparison to the ventricle, the atrial tissue has (1) a more negative sodium channel half-inactivation voltage (V0.5; inactivation curve shifted negatively), (2) a more depolarized RMP, and (3) a slower phase 3 repolarization and therefore shorter diastolic interval (DI), leading to reduced time for sodium channels to recover from inactivation. It has also been suggested that there exist tissue-specific isoforms of the cardiac sodium channel and/or differences in auxiliary subunits, which could lead to the variation in biophysical channel properties observed between atrial and ventricular myocytes. 8,120 Additionally, the shape of the atrial AP is thought to promote greater ranolazine binding, especially at rapid stimulation rates, where DI is reduced or even eliminated and takeoff potentials are more positive.
Heart rate is an important determinant of INa block by compounds, particularly those with rapid unbinding kinetics, such as ranolazine and amiodarone: the faster the rate, the greater the block. 9 At slow heart rates, ranolazine block is more selective for INa,late versus INa,peak, such that APD may be abbreviated by ranolazine without effecting Vmax or excitability. At rapid pacing rates, although there is an increase in the potency of INa,late block by ranolazine, the absolute amount of block decreases, due to a reduction in INa,late density (“available INa,late”) and abbreviated APD. The consequence of this is at higher pacing rates, ranolazine loses its selectivity for blocking INa,late and block of INa,peak becomes more prominent, that is, reduction in Vmax and excitability. 9
The protection offered by ranolazine against ventricular arrhythmias occurs despite its IKr blocking properties and QT interval prolonging effects. This is thought to be due to the accompanying block of INa,late. 114 There exists a TDR in the heart, due to differing levels of ion channels, whereby M cells generally exhibit longer APD than epicardial cells. Alterations in ion currents that increase TDR can act as a substrate for the initiation of arrhythmias. Due to heterogeneity in ion channel density, application of ranolazine decreases APD in M cells (predominant INa,late inhibition) and increases APD in epicardial cells (predominant IKr inhibition), thereby reducing, or not affecting, TDR. 121 As previously mentioned, INa,late is upregulated in several cardiac pathologies. The inhibitory effect of ranolazine on INa,late in both atrial and ventricular tissue is thought to be protective against sodium and calcium overload. Avoidance of sodium and calcium overload would certainly reduce the risk for triggered activity. Indeed, our group has recently shown that ranolazine reduces SR Ca2+ leak in atrial cardiac myocytes isolated from patients with AF. 35 Furthermore, ranolazine has been shown to improve diastolic function, 17 and this is likely due to its effect in limiting cytosolic Ca2+ accumulation. 122 Reduced diastolic wall tension improves coronary blood flow and is thus protective against ischemic events. It has been shown that ranolazine protects against INa, late-induced APD prolongation, and furthermore, it suppresses EADs and ventricular tachycardias. 38,39 In summary, current data suggest that the multichannel blocking properties of ranolazine protect against arrhythmia in both the atria and the ventricles by (1) predominantly INa,peak and IKr synergistic block in the atria, producing PRR and reducing excitability and (2) INa,late and maybe also IKr block in ventricle, reducing TDR and protecting against proarrhythmic mechanisms such as triggered activity. Furthermore, inhibition of INa,late in the atrial and ventricular tissue protects against sodium–calcium overload. Importantly, block of INa,peak by ranolazine appears to be relatively atrial selective, and therefore, excitability is not affected in the ventricles. The effects of ranolazine in atrial and ventricular tissue are summarized in Figure 3.
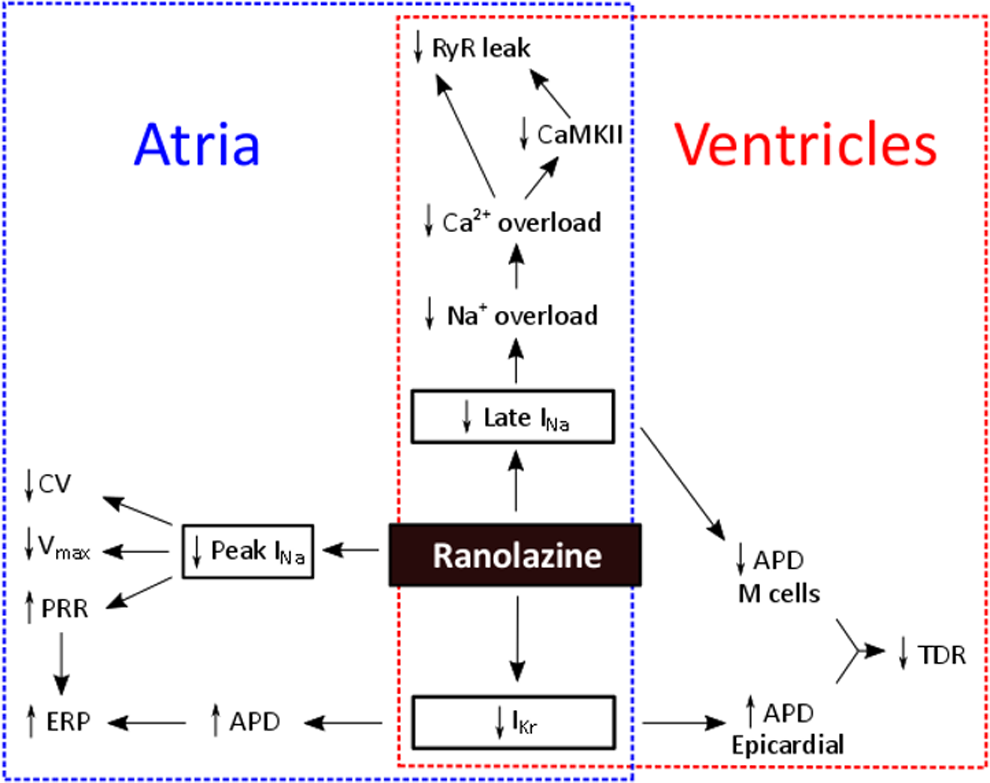
The electrophysiological effects of ranolazine in the atrial and ventricular tissue. Ranolazine block of INa,late protects against arrhythmogenic activity in the atrial and ventricular tissue by reducing intracellular sodium–calcium overload. Concurrent inhibition of INa,late and IKr in ventricular tissue reduces transmural dispersion of repolarization (due to relative channel densities across the ventricular wall) and thus arrhythmogenic risk. Ranolazine increases effective refractory period (ERP) in atrial tissue, constituting an antiarrhythmic effect, by increasing APD (IKr block) and by inhibition of the peak Na+ current (INa,peak) that reduces excitability (decreased Vmax) and conduction velocity (CV).
Ranolazine and amiodarone have been shown to act synergistically in the suppression of pulmonary vein triggered activity, prevention of AF induction, or conversion of recent-onset AF in experimental models. 123,124 Such findings have led to the idea that a multidrug approach for the treatment of AF may hold considerable potential. Dronedarone is a noniodinated derivative of amiodarone with a more favorable safety profile. The combination of ranolazine and dronedarone has been shown to synergistically protect against and suppress AF and to reduce vulnerability to ventricular arrhythmia. 71,72 Furthermore, results from the recent HARMONY trial, a phase II study, show that the combination of ranolazine and low-dose dronedarone reduces AF burden in patients with paroxysmal AF. 5
Currently being developed are highly selective INa,late blockers without accompanying inhibition of IKr. This pharmacological profile could prove effective against arrhythmia development in the ventricle by preventing QT prolongation. One such drug is GS-458967, which has been shown to reduce APD and abolish triggered activity (induced by ATX-II or IKr block) in canine Purkinje fibers, pulmonary vein, and superior vena cava sleeve preparations. 125 Another selective INa,late inhibitor, GS-6615, is currently undergoing a clinical trial (TEMPO trial) in participants with an implanted cardioverter defibrillator, where ventricular arrhythmia and shock occurrence will be assessed. Although these compounds may be promising antiarrhythmic therapies in the ventricle, a multichannel blocking approach, as in the case of ranolazine, may be a more appropriate treatment for AF, for the electrophysiological reasons discussed above.
Clinical Perspective and Conclusions
Heart failure and cardiac arrhythmias are major causes of death in the developed world, and this situation is compounded by an aging population. Identifying safe and effective treatments for arrhythmias in patients with structural heart disease, particularly in cases where AF is also present, poses a considerable challenge. The use of amiodarone for the treatment of arrhythmias, both atrial and ventricular, is accompanied by considerable risk of severe side effects and also bradyarrhythmias. Thus, the discovery of antiarrhythmic agents with a more favorable safety profile, particularly for the treatment of AF, would represent a major advance in medicine. The antiarrhythmic capacity and safety of ranolazine use were demonstrated in the MERLIN-TIMI 36 trial, 3,4,113 but it should be taken into consideration that the initial intravenous administration used in this study differs from the normal daily practice of oral intake. The randomized Ranolazine in Atrial Fibrillation Following An ELectricaL CardiOversion (RAFFAELLO) trial investigated ranolazine in the prevention of AF recurrence after electrical cardioversion, with the aim of determining appropriate dosing. 2 Ranolazine is routinely used at 500 to 1000 mg oral doses twice daily (recommended maximum dose in United States: 1000 mg, Europe: 750 mg), reaching a steady state in the body within 3 days, and over the concentration range 0.25 to 10 μg/mL, ranolazine is 62% bound to human plasma proteins. A lower dose (375 mg) can initially be administered, but in light of the findings of the RAFFAELLO trial, the lower dose may not be sufficient to provide protection against arrhythmias. 2
INa,late has been shown to be increased in several cardiac pathologies, including those highlighted above: structural heart disease and AF. Therefore, patients having ischemic heart disease and/or heart failure possibly associated with paroxysmal AF may especially benefit from the inhibition of INa,late. Increased INa,late is thought to lead to intracellular sodium ion accumulation and thence onto perturbations in Ca2+ handling. INa,late block by ranolazine is therefore thought to protect against these events and associated arrhythmogenic activity. Finally, and perhaps, the key factor contributing to the promising safety profile of ranolazine is the fact that ranolazine block of peak INa is relatively selective to atrial tissue. This property causes a reduction in excitability and CV in the atria (anti-AF effects), without producing deleterious effects in the ventricle, where excitability is maintained. Despite the promising experimental and clinical data gathered thus far, randomized trials of ranolazine use, alone and in combination with other agents, are ultimately required to assess the real antiarrhythmic potential of ranolazine in the clinic; if successful, ranolazine may indeed represent new hope in the treatment of cardiac arrhythmias, especially in patients with structural heart disease.
Footnotes
Author Contributions
F. Mason contributed to design, contributed to interpretation, and drafted the manuscript. S. Sossalla contributed to conception and design, contributed to interpretation, drafted the manuscript, critically revised the manuscript, gave final approval, and agrees to be accountable for all aspects of the work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Sossalla receives speaker’s honoraria from Berlin-Chemie.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Both authors are funded by the Deutsche Forschungsgemeinschaft (DFG) through the Sonderforschungsbereich (SFB) 1002 (A03).