
Abstract
Ranolazine (Ran) is a novel anti-ischemic agent with electrophysiologic properties mainly attributed to the inhibition of late Na+ current and atrial-selective early Na+ current. However, there are only limited data regarding its efficacy and mechanism of action against atrial flutter (Afl) and atrial fibrillation (AF) in intact animals. Therefore, we aimed to investigate the electrophysiologic mechanism of Ran in a rabbit model of inducible atrial tachyarrhythmias elicited by acetylcholine (ACh). Arrhythmias were produced in 19 rabbits by rapid atrial burst pacing during control, after intravenous ACh and after Ran + ACh administration. Recording of right atrial monophasic action potentials (MAPs) and programmed stimulation were utilized to determine the duration of atrial repolarization at various cycle lengths and voltage levels of action potential, including 75% of total MAP duration (MAPD75), effective refractory period (ERP), and postrepolarization refractoriness (PRR = ERP − MAPD75) prior to and after Ran. Control stimulation yielded no arrhythmias or maximal nonsustained runs of Afl/AF. Upon ACh, 17 of 19 rabbits exhibited sustained Afl and AF as well as mixed forms of Afl/AF, while 2 animals revealed none or short runs of nonsustained arrhythmias and were excluded from the study. High-frequency burst pacing during the first 30 minutes after Ran + ACh failed to induce any arrhythmia in 13 of 17 rabbits (76%), while 2 animals displayed sustained Afl/AF and 2 other animals nonsustained Afl/AF. At basic stimulation cycle length of 250 milliseconds, Ran prolonged baseline atrial ERP (80 ± 8 vs 120 ± 9 milliseconds, P < .001) much more than MAPD75 (65 ± 7 vs 85 ± 7 milliseconds, P < .001), leading to atrial PRR which was more pronounced after Ran compared with control measurements (35 ± 11 vs 15 ± 10 milliseconds, P < .001). This in vivo study demonstrates that Ran exerts antiarrhythmic activity by suppressing inducibility of ACh-mediated Afl/AF in intact rabbits. Its action may predominantly be related to a significant increase in atrial PRR, resulting in depressed electrical excitability and impediment of arrhythmia initiation.
Keywords
Introduction
Ranolazine (Ran) is a new antianginal drug recently approved for clinical use with additional electrophysiologic properties on both the atrial and ventricular level. It has been suggested to be a strongly atrial-selective inactivated-state sodium channel blocker 1 in contrast to open-state sodium channel blockers, such as propafenone, which have been demonstrated to act equally on both atria and ventricles. 2 More recent publications, however, indicated that Ran is an open-state blocker that unbinds from closed Na+ channels unusually fast but is trapped in the inactivated state. 3 –5 In vitro investigations demonstrated that at therapeutically relevant concentrations, Ran mainly produces a use-dependent inhibition of late Na+ current (INaL) and atrial-selective peak (transient) INa, as well as a blockade of the rapid component of delayed rectifier K+ current (IKr). 6 Its IKr-blocking effect is attenuated by inhibition of INaL and, therefore, it would be expected that Ran causes minor net changes in atrial repolarization.
The INa inhibition per se suppresses excitability during the late repolarization phase and increases refractoriness independent of changes in repolarization. This mode of action characterizes drugs showing postrepolarization refractoriness (PRR) 7 –9 as the prevailing antiarrhythmic mechanism. Furthermore, Ran at therapeutic doses showed a strikingly dominant atrial selectivity in blocking INa in vitro. 6 Accordingly, it prolonged the final phase of repolarization in normal atrial but not in normal ventricular myocytes, whereas the much less atrial-selective inactivated-state Na+ channel blocker lidocaine shortened repolarization in both cell types.
Acetylcholine (ACh) is known to abbreviate atrial repolarization and refractoriness via activation of the atrial muscarinic-activated potassium current (IKACh) and to facilitate induction of atrial flutter (Afl)/atrial fibrillation (AF). 10 Moreover, ACh has been shown to act more prominently in atrial endocardium than in epicardium thereby increasing dispersion of repolarization and promoting intra-atrial reentry. 11 On the other hand, because intracellular Ca2+ overload secondary to an increased INaL has been found to generate triggered activity arrhythmias, 12,13 inhibition of late Na+ and Ca2+ currents with Ran could abolish such arrhythmias by depressing early and delayed afterdepolarizations.
Since there still exist limited data proving that Ran is an established antiarrhythmic drug, this study aims to add evidence regarding the efficiency of Ran to prevent inducibility of Afl/AF and to elucidate its electrophysiologic mechanism in an in vivo rabbit heart model.
Methods
Experimental Preparation and Recording Procedure
Animal care conformed to international guidelines. A total of 23 New Zealand White rabbits of either sex and mean body weight of 3.6 ± 0.4 kg were studied. Surgical procedures were performed after sedation with 2.5 mg/kg midazolam + 25 mg/kg ketamine intramuscularly (IM), followed by slow intravenous (IV) bolus injection of 15 mg/kg thiopental sodium via the marginal ear vein to induce sufficient anesthesia. Supplemental doses were given as needed in order to maintain surgical anesthesia during the entire experiment. Butorphanol 0.1 mg/kg IM was routinely given for additional analgesia. A catheter was placed in the vena cava (via the left external jugular vein) for administration of fluids along with investigational drugs. Intubation was achieved via tracheotomy, and rabbits were ventilated with a mixture of room air and oxygen at a rate of 30 to 35 cycles per minute and a stroke volume of approximately 8 to 10 mL/kg. For that purpose, a constant-cycled respirator for large animals (Harvard Apparatus Ventilator Model 613, Holliston, Massachusetts) was used. Body temperature was held close to 38°C utilizing a heating pad.
A limb lead electrocardiogram (ECG) was monitored from subcutaneous needle electrodes along with arterial blood pressure by means of a catheter positioned in the aortic arch via the left common carotid artery. Endocardial monophasic action potentials (MAPs) were recorded with a combination contact MAP-PACING electrode catheter (Model 1675P, EP Technologies, Sunnyvale, California) advanced via the right external jugular vein into the right atrium under fluoroscopic guidance (Model 1675P). Proper contact of the tip electrode with the endocardium was recognized by the display of MAP signals. Adequate pressure of the tip electrode against the endocardium could be determined by the quality of the signal received. To avoid direct current (DC) drift, the MAP proximal and distal electrodes were shorted and soaked in saline for 30 minutes prior each experimental use. All signals were displayed on a multichannel monitor oscilloscope (Nihon Kohden RM-6000 polygraph amplifier system) equipped with a thermal array recorder (WS-682G). The MAP signals were amplified by high input impedance, MAP DC-coupled isolated, differential preamplifier (AB-601 G by Nihon Kohden). The frequency response of the MAP recording system was from DC to 5000 Hz.
Electrophysiologic Protocol
All animals studied were subjected to 2 types of atrial stimulation, programmed stimulation (repetitive extrastimulation) in order to determine atrial action potential duration (MAPD) and effective refractory period (ERP), as well as high-frequency burst atrial pacing to elicit sustained Afl/AF. For that purpose, a Grass stimulator (Model S8800, Quincy, Massachusetts) was used to deliver constant-current rectangular stimuli for high fixed-rate pacing or for delivering premature stimuli at progressively decreased coupling intervals. Pacing stimuli were of 2-millisecond duration at twice diastolic threshold intensity which was checked repetitively during the stimulation protocol. The atrium was paced at basic drive cycle lengths of 250, 200, and 150 milliseconds that did not allow capturing of sinus beats to determine MAPDs at 30%, 50%, and 75% repolarization (MAPD30, MAPD50, and MAPD75) and ERP in all pacing cycle lengths (PCLs), respectively. The MAPD was determined as the interval from the time of MAP depolarization to the respective level of repolarization. The ERP was determined with 5 milliseconds precision and defined as the longest coupling interval between the last basic drive stimulus and the extrastimulus that failed to elicit a propagated response. Postrepolarization refractoriness was measured manually as ERP minus MAPD75 during premature extrastimulation in the MAP recording that was used for pacing at a paper speed of 100 mm/s.
After determining the basic electrophysiologic variables by programmed atrial stimulation, high-frequency burst pacing was used to provoke arrhythmias during control settings pre- and post-ACh infusion (0.02 mg/kg per min IV). This is equivalent with 10−5 mol/L ACh (approx 1.82 mg ACh). This amount of ACh was dissolved in 100 mL of 0.9% NaCl solution and the perfusion was set at 100 to 200 mL/h in order to induce sustained arrhythmia. Then, Ran was administered (2.4 mg/kg IV bolus followed by 0.135 mg/kg per min continuous infusion), basic electrophysiologic parameters were reevaluated, and ACh was applied again parallel to Ran to test the arrhythmia reproducibility. Each arrhythmia was reproducibly induced for at least 2 times during baseline and after drug administration. In cases of post-Ran sustained arrhythmia reproducibility, the rate of the respective arrhythmia was assessed and compared to predrug values. The Afl/AF induction was attempted by burst atrial pacing initially at twice diastolic threshold and then by increasing the threshold to 3 times or more to reduce PRR and thereby to enable capture at earlier repolarization levels. This technique for arrhythmia induction was used for burst PCLs started from 200 milliseconds and decreased stepwise by initially 10 milliseconds and then near the ERP by ≤5 milliseconds intervals up to 60 milliseconds or shorter PCLs. Each attempt consisted of 5 to 8 beats while the recovery time between 2 consecutive attempts was 10 to 15 seconds. The end point of pacing protocol was the elicitation and reproducibility of sustained (≥ 30 seconds) Afl and/or AF.
Atrial fibrillation was defined as a totally irregular atrial rhythm with an average cycle length <150 milliseconds, whereas Afl was defined as a rapid but regular atrial rhythm with fixed cycle length >150 milliseconds. Arrhythmia definitions were made based on atrial MAP configuration and not on patterns of surface ECG signals. Both forms of arrhythmia occurring separately or showing spontaneous transition of the one form into the other were defined as mixed Afl/AF. Termination of sustained Afl/AF occurred either spontaneously after 30 seconds or by stopping ACh infusion. The dosing scheme and the infusion rate of ACh were so balanced to achieve sustenance of the arrhythmia by avoiding total atrioventricular block or lethal hypotension.
Both ACh and Ran were purchased from Sigma-Aldrich (Germany), dissolved in distilled water and prepared fresh before each experiment.
Statistical Analysis
Electrophysiologic variables were expressed as mean ± standard deviation (SD). Comparisons of results between baseline and drug data were performed using paired Student t test. For categorical variables, such as the reinitiation of sustained Afl/AF pre- and post-Ran, the McNemar test on paired proportions was used. Statistical significance was assumed at P value <.05.
Results
From the 23 animals initially included in the study, 2 animals succumbed to interventions related to anesthesia while 2 other animals showed irreversible hypotension and cardiac arrest in response to ACh. Of the remaining 19 animals, 2 revealed nonsustained atrial arrhythmias when exposed to ACh and were excluded from the study. Finally, a total of 17 rabbits were adequately investigated.
Ranolazine increased atrial action potential duration at 30%, 50%, and 75% repolarization compared with control measurements during spontaneous sinus rhythm (Figure 1), and upon stable atrial pacing (Figure 2). These differences remained statistically significant also at higher atrial pacing rates. Programmed atrial stimulation at basic drive (S-S) pacing cycle lengths (PCLs) of 250, 200, and 150 milliseconds and single extrastimuli (S-S1) was used to determine the electrophysiologic parameters summarized in Figure 2. Ranolazine caused more pronounced increases in atrial refractoriness than repolarization leading to the appearance of PRR. Pacing thresholds were verified throughout the protocol and remained stable in order to minimize artificial PRR caused by changes in pacing threshold. Positive values of PRR in control experiments reflect that reexcitability occurred at a repolarization level exceeding MAPD75.

Effects of ranolazine on heart rate and atrial MAPD during sinus rhythm in rabbit. Note a characteristic slowing in heart rate, diastolic period, and a moderate prolongation of MAPD in comparison to control electrograms. MAPD indicates total monophasic action potential duration.

Effects of Ran on MAPD at 30%, 50%, and 75% repolarization, ERP, and PRR in vivo. Ran increased PRR at all PCLs (250-150 milliseconds) significantly and thus retained its antiarrhythmic activity at higher atrial rates. The relative difference between control PRR and Ran PRR remained stable at basic drive PCLs from 250 to 150 milliseconds. ERP indicates effective refractory period; MAPD, total monophasic action potential duration; PCL, pacing cycle length; PRR, postrepolarization refractoriness; Ran, ranolazine.
High rate burst pacing elicited sustained and reproducible Afl and AF in the majority of animals, 1 to 2 minutes after exposure to ACh (Figure 3). Most of these arrhythmias were induced by pacing intervals ranging between 80 and 130 milliseconds, namely close the ERP. Using the same pacing protocol, no significant arrhythmias were inducible during control conditions. Acetylcholine transiently reduced the arterial blood pressure accompanying by mild-to-moderate ischemic alterations, as indicated by ST-segment depression in surface ECG leads. The fact that cessation of ACh infusion during a sustained arrhythmia led to its relatively abrupt termination suggests strong dependence of arrhythmia persistence from drug concentration. The time interval between spontaneous termination of arrhythmia and cessation of ACh infusion averaged 10 to 15 seconds.

This figure summarizes the absolute number of animals exhibiting sustained or nonsustained Afl/AF (sAfl/AF or nsAfl/AF) in the control, during ACh, and after Ran + ACh using burst pacing. See text for details. ACh indicates acetylcholine; AF, atrial fibrillation; Afl, atrial flutter; nsAfl, nonsustained Afl; Ran, ranolazine; sAfl, sustained Afl.
After Ran administration, all electrophysiologic variables were reevaluated before ACh was administered concomitantly with Ran to test the reinducibility of arrhythmia by atrial burst pacing. During the first 30 minutes following Ran administration, ACh plus burst pacing revealed no arrhythmias in 13 of 17 (76%) animals (Figure 3). The 4 animals displaying reinducible sustained or nonsustained Afl episodes even after Ran administration, revealed apparently longer arrhythmia cycle length intervals compared with pre-Ran values (80 ± 10 to 95 ± 12 milliseconds, pre- vs post-Ran). Also reinducible AF post-Ran consisted of lower frequency waves than those observed in pre-Ran episodes of AF. With special reference to sustained Afl/AF, Ran significantly abolished reinitiation of arrhythmia in 15 of 17 animals (χ2 = 18.733, df = 1, P < .001).
Figures 4 and 5 are representative examples illustrating the effects of ACh on sinus cycle length, arterial blood pressure, right atrial MAP duration (RAMAPD), and arrhythmogenesis. Acetylcholine slowed the heart rate with moderate decreases in blood pressure at the beginning of infusion, shortened RAMAPD, and facilitated the inducibility of AF/Afl compared with the control. Ranolazine inhibited reproducibility of these arrhythmias despite continuation of ACh administration.
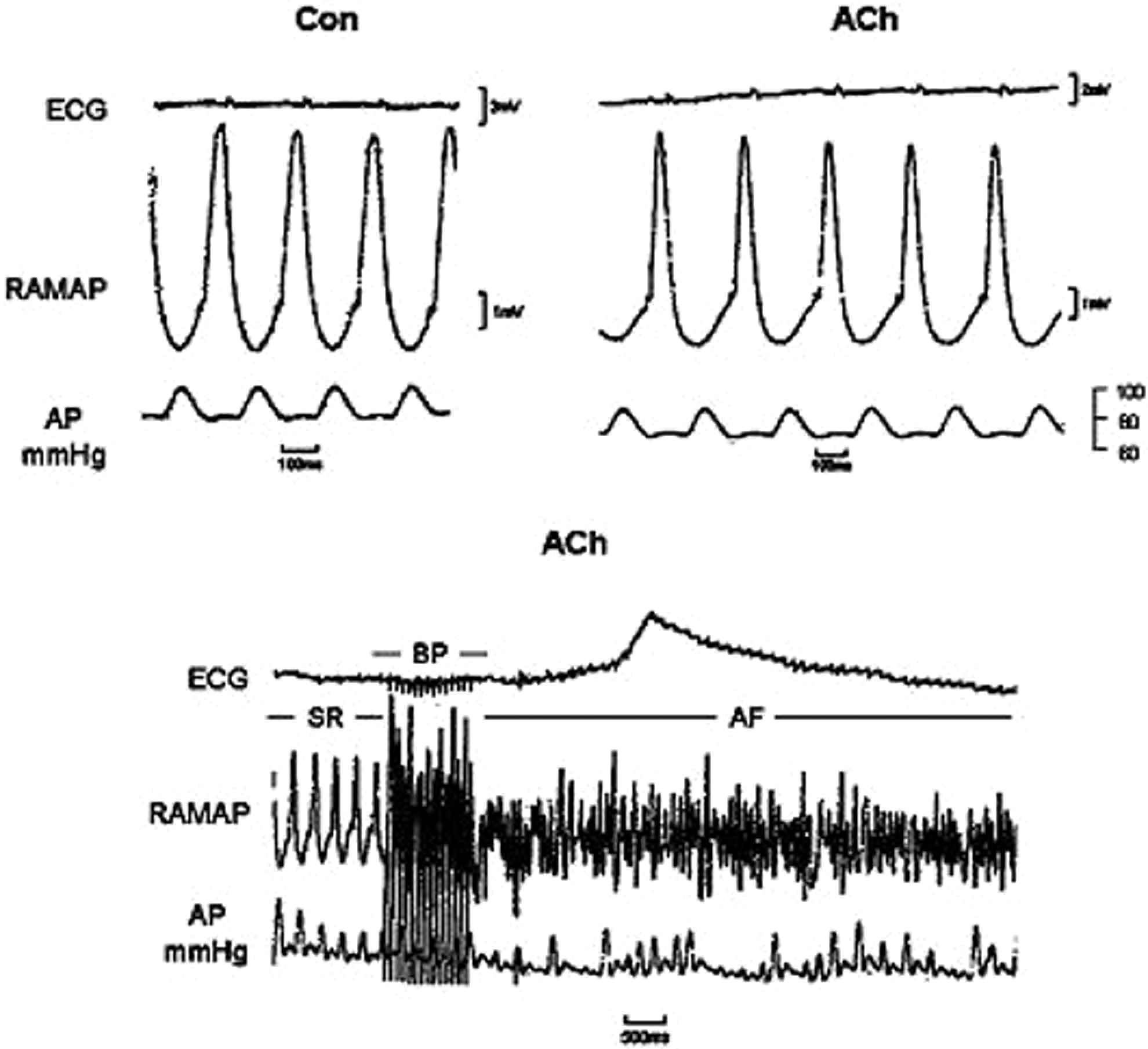
This figure shows a typical experiment at which ACh causes simultaneous slowing in heart rate, while decreased arterial pressure and shortened atrial repolarization (top recordings). Bottom recordings show the induction of AF with rapid burst pacing (BP). SR indicates sinus rhythm; AP, arterial pressure; ACh, acetylcholine; AF, atrial fibrillation.
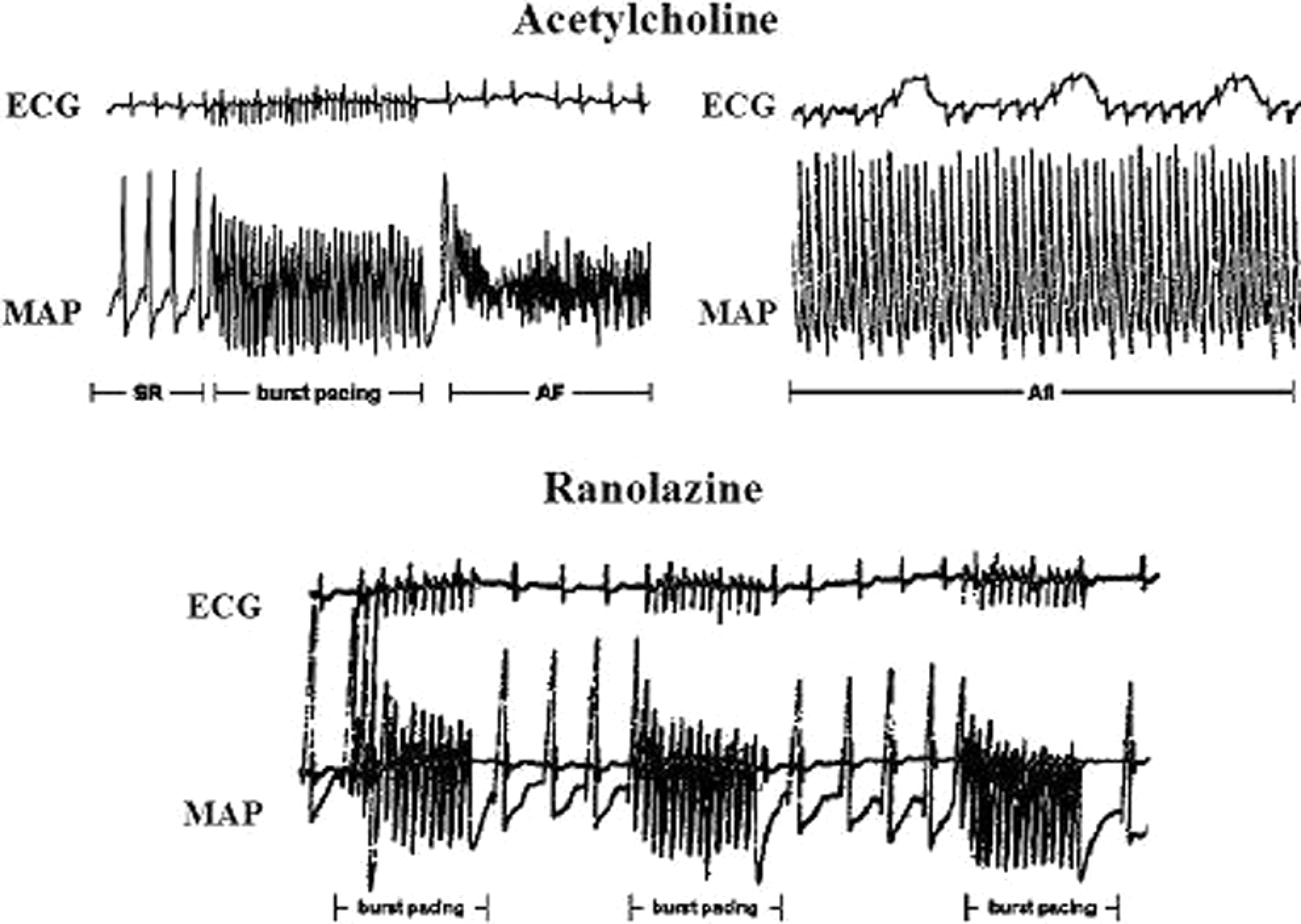
Representative episodes of AF (left top corner) and Afl (right top corner) were reproducibly induced after ACh administration and burst pacing. These arrhythmias were frequently induced in the same animal. After Ran, successive runs of burst pacing did not induce Afl or AF despite concomitant ACh administration. Note the differences in MAP shape during local AF and Afl, and the smaller decreases in MAP duration in the bottom tracings with Ran plus ACh. ACh indicates acetylcholine; AF, atrial fibrillation; Afl, atrial flutter; MAP, monophasic action potential; Ran, ranolazine.
The following standard ECG variables were obtained 10 minutes after Ran in the absence of ACh (mean ± SD, control vs Ran, n = 17): sinus cycle length 312 ± 41 versus 351 ± 56 milliseconds (P = .027), PQ interval 75 ± 12 versus 88 ± 14 milliseconds (P = .007), QRS complex 45 ± 5 versus 47 ± 7 milliseconds (P = .345), and QT interval 140 ± 11 versus 150 ± 14 milliseconds (P = .027) determined during stable right atrial pacing at 300 milliseconds of cycle length.
Discussion
This study investigated the antiarrhythmic mechanism of the novel antianginal agent Ran on ACh-induced Afl/AF in intact rabbits. Ranolazine caused a significant increase in right atrial PRR and suppressed Afl/AF inducibility equally in 76% of all animals exposed to ACh, thereby confirming in vitro data by Burashnikov et al in canine right atrial preparations. 6 The PRR prolongation after Ran might hinder premature atrial extrastimuli to capture repolarization and to promote arrhythmogenesis. The effect of ACh to abbreviate atrial repolarization and refractoriness underlies its ability to facilitate the induction of atrial arrhythmias. Ran blocks IKr, thereby contributing to the prolongation of atrial repolarization under these conditions. However, the increase in ERP and development of PRR are principally due to the actions of the drug to block INa. These effects of Ran blunt the marked ACh-induced abbreviation of repolarization and underlie its antiarrhythmic efficacy in this setting.
Recent experiments in pigs demonstrated an antiarrhythmic effect of Ran, as evidenced by increasing atrial refractoriness, suppressing ACh-induced AF, and by substantially reducing arrhythmia duration by utilizing bipolar intracardiac electrograms. 14 In our experiments, we used MAPs that enable more precise measurement of local repolarization and refractoriness which are needed to calculate PRR. These investigators reported that Afl was never inducible in the control state during their experiments, thus Ran could not be tested against Afl. Whether high myocardial ACh concentration, possibly achieved by the intrapericardial application method, is an explanation for this finding is not clear. In our model, both Afl and AF were reliably induced with right atrial burst pacing shortly after starting with IV ACh infusion. This is consistent with the findings reported by Waldo revealing that Afl is a macroreentry phenomenon almost always located in the right atrium. 15
In these experiments, persistent Afl/AF was not inducible during control conditions despite successive stimuli trains of maximal strength and very short cycle length. Continuous infusion of ACh at 0.02 mg/kg per min or higher enhanced atrial susceptibility to sustained flutter and/or fibrillation. Because ACh has no effect on atrial conduction, it seems likely that these arrhythmias were strongly dependent on the ACh-induced shortening in atrial repolarization and refractoriness, due to activation of IKACh and, probably, of slow inward calcium current ICa. 16,17 This shortening of atrial repolarization permits very rapid rates of excitation that might induce intracellular calcium overload and afterpotentials at more incomplete repolarization levels of action potential. Such a mechanism causing phase 3 early afterdepolarizations (EADs) and atrial extrasystoles after rapid pacing has been described. 18 Moreover, the interplay between ACh and calcium has been confirmed by Wang et al 19 who demonstrated that withdrawal of ACh elicited a cyclic adenosine monophosphate-mediated rebound stimulation of L-type Ca2+ current and Na+/Ca2+ exchange current, resulting in delayed afterdepolarizations (DADs).
In our study, ACh alone did not result in the appearance of any form of afterpotentials during sinus rhythm, except of slowing in heart rate and marked abbreviation in atrial MAP duration. This observation supports earlier findings by Allessie et al showing that intra-atrial reentry was the basic underlying mechanism of ACh-induced Afl (one circuit present) and possibly AF (multiple circuits concept) in the dog. 10 In this concern, in vitro investigations in canine atrial cells demonstrated that ACh shortened right atrial repolarization more prominently in endocardium than in epicardium, 11 thereby increasing spatial dispersion of repolarization perpendicular to the atrial wall. Parallel to this, release of endothelin and nitric oxide in response to muscarinic stimulation from endocardial endothelium might accentuate the abbreviating effect of ACh on endocardial repolarization by activating IKACh and inhibiting ICa. 20 Obviously, these data cannot be extrapolated to all animal species and experimental protocols employed by different investigators. For example, Wang et al 21 found that Ito blockade significantly increased epicardial but not endocardial repolarization in guinea pig atrial cells, whereas other authors 11 showed that Ito blockade had similar effects in canine atrial epicardium and endocardium.
Ranolazine is not an ACh antagonist but it exerts electrophysiologic effects that abrogated those of ACh in atrial myocardium. Accordingly, Ran exerts opposite effects to IKACh activation via inhibition of IKr, thereby preventing shortening of ACh-induced atrial repolarization. In addition, Ran inhibits INaL which is very small in normal cardiac cells and for this reason, inhibition of this current did not completely offset the prolongation of action potential induced by IKr blockade. This might result in a net moderate prolongation of atrial action potential duration. Moreover, Ran led to a marked increase in atrial refractory period exceeding the increase in repolarization. This mechanism of action, called PRR, can protect the heart against arrhythmias by rendering it refractory to extrasystoles falling during the vulnerable period, namely close the ERP.
The fact that inhibition of Ca2+ release from the sarcoplasmic reticulum by ryanodine suppressed DADs but not EADs, 22 may give a putative role to INaL for a contribution to triggering arrhythmias. In this respect, ATX-II induced increase in INaL may cause afterpotentials directly by generating EADs or indirectly by inducing DADs via an increased sodium-dependent calcium loading at early diastole. Reduction in INaL would, therefore, be expected to suppress EADs and to cancel sodium-induced reverse-mode sodium-calcium exchanger (NCX) activation which is responsible for calcium-overload-induced DADs. 13,23
Sustained activity can result from channels that fail to inactivate or channels that recover from inactivation during repolarization. Rajamani et al showed that Ran caused a significant use-dependent slowing of the recovery from inactivation of peak INa, suggesting an interaction of the drug with inactivated states of the channels. 24 The prolongation of IKr-induced action potential potentiates the effect of Ran to produce inactivated-state block of the sodium channel during repolarization, thereby making sodium channels less available and more sensitive to block by Ran. This could explain the prolongation of atrial refractoriness much more than repolarization after Ran causing PRR, because the repolarization lengthening effect of Ran is mitigated by concomitant inhibition of INaL. However, more recent data demonstrated that Ran is a predominantly open-state sodium channel blocker, staying trapped in the pore of the channel during inactivation and unbinding during the preopen/resting state. 3 –5 Accordingly, for the inactivated voltage-gated sodium channel (rNav1.4) Na+ channel block, Ran is weaker than lidocaine and block of open Na+ channels is via the conserved local anesthetic receptor with a relatively slow on-rate.
Special consideration should be given to reports by Belardinelli et al emphasizing that at clinically relevant concentrations, Ran is selective for the block of INaL in a 38:1 ratio over peak INa. 25 Similar to lidocaine, Ran blocks Na+ channels in the inactivated state in dogs with heart failure 1 with an apparent increased sensitivity of atrial myocytes over ventricular myocytes. Using a canine model, Burashnikov et al demonstrated that Ran exhibits use-dependent block of Na+ channels and prolongation of repolarization in atrial but not in ventricular myocytes. 6
Positive PRR values in control conditions might partially be due to the relative low diastolic threshold intensity of pacing impulses. Similarly, Kirchhof et al found positive predrug PRR values at ventricular level which decreased with increasing stimulus strength. 26 They suggested that decreased PRR promoted encroachment of premature stimuli onto progressively earlier repolarization phases and facilitated arrhythmia induction. This is a reason why we also used high rate burst pacing at maximal stimulus strengths to induce arrhythmias during all steps of our experimental protocol.
Dependent on the stimulation protocol and the animal model used, small positive or negative PRR values can be measured during control states. In our study, it was apparent that atrial excitability assessed during control stimulation recurred at a repolarization level beyond 75%, at which, PRR would be near zero. It should be emphasized, however, that the relative PRR changes determined during control conditions and after drug administration might be more important for the antiarrhythmic potency of a drug.
Limitations
This study was performed in a rabbit model that did not permit a wide specter of pacing rates as in the human heart. Rabbits have normally very high heart rates compared with dogs and pigs, and this limits the possibility to assess PRR at PCLs longer than 250 milliseconds for determining use-dependence of PRR over a wide-range frequency series. Furthermore, it is important to note that we examined animals with intact hearts and very likely at ACh concentrations higher than those encountering in clinical practice. Besides, the arrhythmias observed were not spontaneous but induced with rapid stimulation, possibly depending on mechanisms other than those described in this study. For these reasons, although our findings cannot directly be extrapolated to the human situation, the cholinergic component is known to play a significant role in the genesis of Afl/AF in structurally normal or diseased human heart. On the other hand, reduction in blood pressure and ST-segment depression during higher ACh doses cannot exclude an involvement of transient ischemia as an additional arrhythmogenic factor to the direct electrophysiologic effects of ACh. However, these hemodynamic and ischemia-induced actions of ACh, albeit also present after Ran, did not abrogate the antiarrhythmic effect of Ran in our study.
Clinical Implications
These experiments demonstrate a marked antiarrhythmic action of Ran, as indicated by increasing PRR and preventing the induction of Afl/AF elicited by ACh and premature excitations. Even at higher pacing rates, Ran retained its capacity to prolong PRR and to achieve protection against arrhythmia initiation. This is clinically relevant because the nature of these arrhythmias is characterized by very fast atrial responses and the drug used should maintain protection at these rates. Based on our findings, lengthening of refractoriness not related to lengthening of repolarization might theoretically function also in the diseased heart, particularly when a prolongation of repolarization is undesirable. Inhibition of inward currents during early and late repolarization might protect the electrical stability of the heart particularly when a disease or a variety of QT-prolonging drugs induce abnormal prolongation of repolarization. This study suggests that Ran possesses significant antiarrhythmic activity against Afl/AF and may be useful in patients with atrial tachyarrhythmias occurring in conjunction with coronary artery disease and after cardiac surgery (postoperative Afl/AF). Our results and a comprehensive review by Antzelevitch et al 27 on the electrophysiologic and hemodynamic properties of Ran tacitly support broading of the indication specter of this agent beyond its current use for chronic angina.
Footnotes
Acknowledgments
We gratefully acknowledge the technical assistance of I. Makantasis and D. Makris (MD, CCA, MRCP) for editing the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.