
Abstract
The peptide hormone relaxin has traditionally been linked to the maternal adaptation of the cardiovascular system during the first trimester of pregnancy. By promoting nitric oxide formation through different molecular signaling events, relaxin has been proposed as a pleiotropic and cardioprotective hormone in the setting of many cardiovascular diseases. In fact, preclinical studies were able to demonstrate that relaxin promotes vasodilatation and angiogenesis, ameliorates ischemia/reperfusion injury, and regulates extracellular matrix turnover and remodeling. In the RELAX-AHF phase 3 clinical trial, serelaxin (recombinant human relaxin) was shown to be safe, and it exerted survival benefits in patients with acute heart failure. RELAX-AHF-2 is currently ongoing, and it aims to address a larger population and evaluate harder clinical outcomes. Besides heart failure, acute myocardial infarction, peripheral arterial disease, and stable coronary disease could be target diseases for treatment with serelaxin in future clinical trials.
Introduction
Despite the advances in cardiovascular medicine, ischemic heart disease continues to be the leading cause of heart failure (HF) worldwide. 1 The therapeutic approaches to reduce injury secondary to acute myocardial infarction (AMI) and HF have improved dramatically as reflected by the enhanced clinical outcomes and quality of life in both populations. 2 However, HF remains a major health concern worldwide, currently affecting approximately 5 million patients in the United States alone and causing more than 1 million hospitalizations each year. 1 Moreover, hospitalization for acute heart failure (AHF) is one the most important predictors of morbidity and mortality among patients with HF, 3 and, unfortunately, readmission and mortality after discharge in patients with HF continue to occur between 10% to 20% and 20% to 30%, respectively, within 3 to 6 months. 4 Therefore, there is an unmet need to develop novel strategies to combat ischemic heart disease and HF.
Since the mid-1990s, basic and translational research have interrogated the cardioprotective role of relaxin and its potential mechanism(s) of action in the cardiovascular system in several animal models. 5 Relaxin has traditionally been related with genital tract adaptation process prior to parturition during pregnancy in lower species. 6 This suggested a key role of relaxin in extracellular matrix (ECM) remodeling and turnover, and further studies confirmed a direct effect of relaxin on ECM. 6 Contrary to lower species, human relaxin plasma levels are highest during the first trimester. 6 Although available data suggest a role of relaxin in hemodynamic adaptation during pregnancy in humans, more clinical evidence in humans is needed. Most studies were conducted in conscious pregnant rats and demonstrated the effect of relaxin on renal vasodilatory actions and cardiovascular adaptations, including reduction in systemic vascular resistance (SVR) and the consequent increase in cardiac output (CO). 7 –9 This evidence has led to hypothesize possible therapeutic targets of this hormone in several diseases such as preeclampsia, scleroderma, renal, and cardiovascular disease. 6
The severity of HF has been correlated with elevated relaxin plasma levels, 10 which implicated a potential therapeutic use of relaxin in patients with HF. Indeed, serelaxin, a human recombinant form of relaxin 2, has been recently tested in RELAX-AHF, a phase III randomized clinical trial in patients with AHF, which demonstrated promising results. 11 Additionally, there is enough evidence to support the premise that relaxin may counteract many pathophysiologic mechanisms that occur during ischemic heart disease. To this end, reperfusion therapy with relaxin in several animal models of ischemia–reperfusion (I/R) injury has shown intriguing cardioprotective results. 12,13
Relaxin: Biology and Mechanisms of Action
Relaxin Peptides and Receptors
The hormone relaxin, 6-kDa polypeptide, is a member of the insulin superfamily. 6 In humans and other primates, 3 relaxin genes have been identified. 6 Relaxin 1 gene is exclusive in these species; however, the expression product human relaxin 1 (H1 relaxin) has not been detected in vivo. 6 The relaxin 2 gene codes for human relaxin 2 (H2 relaxin), which is the major stored and circulating form. Finally, Relaxin 3 gene corresponds and codes for human relaxin 3 (H3 relaxin), which is highly expressed in the brain and has stress-related actions. 6 The human relaxin 2 peptide is the subject of the review and will simply be referred to as relaxin. Relaxin is mostly produced in the corpus luteum and placenta in mammals, however, it can also be produced by other tissues including brain, heart, and kidney. 6
The relaxin receptor 1 (RXFP1), one of four members of the relaxin family peptide receptors, is a G-coupled cognate ligand receptor for relaxin. 5 RXFP1 expression is not only found in a wide range of reproductive tissues but also in nonreproductive tissues including endothelial and smooth muscle cells of arteries and veins, kidney tubules, 14,15 cardiomyocytes and heart tissue, 14,16 lung, liver, blood cells, and brain. 6 Other relaxin receptors and their peptides will not be discussed, since they are not relevant to the aims of this review.
Pleiotropic Signaling Pathways
The hormone relaxin, after coupling with its cognate receptor RXFP1, activates several G-coupled proteins to promote production of cyclic adenosine monophosphate (cAMP), phosphorylation of mitogen-activated protein kinases (MAPKs) and activation of the nitric oxide (NO) pathway 6 (Figure 1). In the acute phase, phosphorylation of protein kinase B (Akt), through phosphatidylinositol 3 kinase (PI3K), activates endothelial nitric oxide synthase (eNOS) for immediate production of NO. 17 Nitric oxide is also produced by inducible NOS (iNOS) in a chronic and sustained manner. 18 The rise in cAMP, after activation of adenylyl cyclase (AC), upregulates protein kinase A (PKA) activity leading to activation of nuclear factor κB (NFκB), a promotor of iNOS, and cAMP response element. 6 Other mechanisms involving activation of NFκB include phosphorylation of ERK 1/2 after activation of Gs. 19 The NFκB also promotes transcription of vascular endothelial growth factor (VEGF) 20 and matrix metalloproteinases. 21 As seen, these pathways triggered by relaxin are complex and involve several effectors and different targets. However, eNOS-driven NO production seems to play a central role in mediating the pleiotropic effects of relaxin.

Central illustration of signaling mechanism(s) of action of relaxin in the cardiovascular system: The combinatory effect of these mechanisms ultimately leads to vasodilatation, antioxidant and anti-inflammatory actions as well as angiogenesis and antifibrotic effects. These effects may prove beneficial in the settings of acute heart failure, acute myocardial infarction as well as chronic conditions such as chronic heart failure, chronic coronary disease, and peripheral arterial disease. Abbreviations: Akt, protein kinase B; AC, adenylyl cyclase; cAMP, cyclic adenosine mono-phosphate; CRE, cAMP response element; eNOS, endothelial nitric oxide synthase; ERK ½, extracellular signaling regulated kinase 1/2; ET-1, Endothelin 1; ETB, Endothelin B Receptor; Gαs, G-coupled protein α-S; Gαi3, G-coupled protein αi3; γ, Subunit γ; β, subunit β; GαOb, G-coupled protein αOb; iNOS, inducible nitric oxide synthase; MMP, matrix metalloproteinase; NFκB, nuclear factor κB; NO, nitric oxide; PI3 K, phosphatidylinositol 3 kinase; PKA, protein kinase A; RXFP1, relaxin receptor-1; VEGF, vascular endothelial growth factor.
Relaxin and the Cardiovascular System
Coupling of relaxin with its RXFP1 receptor causes versatile effects in different cells and tissues as illustrated in Figure 2.

A summary of the protective effects of relaxin in individual cell lines based on experimental studies. Arrows
The Vascular System
Initial clinical studies on relaxin as a therapeutic agent for its vasodilatory properties date back to the 1950s when an impure porcine relaxin was shown to improve signs and symptoms of ischemia in patients with Reynaud syndrome and severe peripheral arterial disease (PAD). 22 Interestingly, in a subset of 3 patients with prior myocardial infarction (MI) and active angina, relaxin was reported to improve symptoms and reduce nitroglycerin requirements. 22 These initial observations suggested that relaxin had, indeed, a direct vasodilatory effect in patients with cardiovascular disease.
In vitro studies with vascular smooth muscle cells have shown that relaxin promotes changes in cell shape and cytoskeleton consistent with cell relaxation, and these effects were associated with increased NO production and reduced cytosolic calcium. 23 Moreover, relaxin was able to increase NO production in rat coronary endothelial cells by upregulating iNOS expression. 24 Interestingly, this indicates that relaxin secreted by the heart may play a key role in dilating human systemic resistance arteries and thus contribute to cardiovascular regulation. 25
Myogenic reactivity, a parameter of vascular tone, was also reduced in isolated small renal and mesenteric rat arteries upon administration of relaxin, 26 suggesting an additional role of relaxin in organs other than kidneys. In this regard, relaxin was able to increase CO and arterial compliance and reduce SVR without altering mean arterial pressure (MAP) in normotensive and hypertensive rats. 27 Chronic administration of relaxin in conscious adult rats increased effective renal plasma flow and glomerular filtration rate (GFR) mediated by NO and attenuated renal vasoconstriction induced by angiotensin II infusion. 28 Observational studies in healthy volunteers also showed that relaxin, after a 5-hour infusion, increased renal plasma flow by 47% compared to basal levels, with no changes in MAP. 29
Concordant with the previous observation in the 3 patients with coronary disease and PAD, whereby relaxin reduced angina and the required nitroglycerin dose, 22 relaxin has shown to be a potent coronary vasodilator. In a study with isolated, perfused rat and guinea pig hearts, relaxin significantly increased coronary blood flow when compared to other vasodilatory agents such as nitroprusside and acetylcholine, through stimulation of NO production. 30
It has also been reported that relaxin possesses proangiogenic effects in both reproductive and nonreproductive organs. In human endometrial cells, relaxin upregulates VEGF in a dose-dependent manner by increasing cAMP. 20 Relaxin also induced VEGF and basic fibroblast growth factor (bFGF) expression in ischemic wound sites. 31 More recently, VEGF inhibitors were reported to prevent the effect of relaxin on myogenic constriction in small renal arteries in rats and human subcutaneous arteries and also abrogated relaxin-induced increase in GFR and effective renal plasma flow in rats, 32 indicating a possible role of VEGF in the vasodilatory actions of relaxin.
Myocardial Ischemia and Reperfusion Injury
The effect of relaxin on myocardial I/R injury was first evaluated in a model of isolated–perfused guinea pig hearts. 33 Relaxin increased coronary flow when given in the perfusion buffer at the time of coronary occlusion. Nitric oxide production was increased with relaxin and, intriguingly, these effects were blunted with NOS inhibition. Additionally, relaxin significantly reduced calcium overload and malonyldialdehyde, markers of myocardial injury and mast cell degradation, as well as mitochondrial swelling and hypercontraction of myofibrils. In a rat model of I/R, pretreatment with relaxin 30 minutes before ischemia reduced myocardial injury, ventricular arrhythmias, and mortality. 13 Furthermore, morphological studies showed that relaxin reduced endothelial and cardiomyocyte swelling and conserved normal ultrastructure of myofilaments, which were also accompanied by reduction in neutrophil accumulation in capillary vessels. These results correlated with reduction in myeloperoxidase activity, a marker of neutrophil accumulation in tissue, and myocardial calcium content. Reperfusion therapy with relaxin following 30 minutes of ischemia in a swine model of myocardial I/R caused dose-dependent reduction in serum biomarkers of myocardial damage. 12,34 These findings correlated with increased myocardial viability measured with cardiac single-photon emission computed tomography with Thallium chloride. Malondialdehyde, calcium overload, and myeloperoxidase were also reduced with relaxin. Moreover, the relaxin-induced reduction in severe ventricular arrhythmias secondary to I/R was associated with mast cell inhibition and a decline in plasma and cardiac content of histamine. 34 In a rat model of MI, relaxin also increased bFGF messenger RNA expression in the peri-infarct region in both cardiomyocytes and fibroblasts, highlighting its proangiogenic benefits following infarction. 31
In vitro studies have evaluated the impact of relaxin on apoptosis. Using cultured neonatal rat cardiomyocytes, relaxin was shown to reduce apoptosis due to oxidative stress induced by hydrogen peroxide. 35 In this study, relaxin-treated cardiomyocytes exhibited augmented Bcl2–Bax ratio, indicative of an antiapoptotic effect, and displayed increased Akt expression. Moreover, relaxin was shown to reduce high glucose-induced apoptosis in neonatal rat ventricular myocytes by suppressing the extrinsic and intrinsic pathways of apoptosis. 36 Recently, relaxin was shown to protect primary mouse cardiomyocytes from hypoxia–reoxygenation injury. 37 Apoptotic cell death was significantly reduced with relaxin, measured by Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, and correlated with an increase in Bcl2–Bax ratio. 37 The effects of relaxin on apoptosis were also assessed in vivo using models of I/R injury, whereby reperfusion therapy with relaxin in a swine model of MI reduced cardiomyocyte apoptosis as measured by caspase 3 activity and TUNEL assay. 12
Relaxin and Myocardial Inflammation
Relaxin also possesses anti-inflammatory traits that contribute to its cardioprotective role against I/R injury, since relaxin treatment attenuated mast cell degranulation as well as neutrophil recruitment and activity. Additionally, relaxin was found to cause a significant reduction in neutrophil adhesion and endothelial expression of adhesion molecules P-selectin and VCAM-1 in vitro using rat coronary endothelial cells after lipopolysaccharide exposure. These findings were blunted with NOS inhibition, suggesting the involvement of NO. 38 Moreover, adhesion was reduced in human and rabbit platelets after treatment with relaxin, which was also shown to be mediated by NO. 39
The role of sterile inflammation that occurs following MI in promoting further injury and the development of HF has been in the spotlight of recent investigations. 40 The NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome is a macromolecular structure that functions as a platform for the production of proinflammatory cytokines of the interleukin 1 family (i.e. IL-1β and IL-18), involved in the progression of several diseases. 41 In the cardiovascular system, it is activated in cardiomyocytes, fibroblasts, leukocytes, and endothelial cells. 40,42 There is enough evidence in animal models regarding NLRP3 activation after myocardial injury 43,44 and related impairment of heart function and remodeling by producing IL-1β and IL-18. 45,46 Although there is no current evidence supporting a modulatory role of relaxin on NLRP3, an inhibitory effect of NO on NLRP3 has been reported. Nitric oxide was shown to inhibit caspase 1, IL-1β, and IL-18 release from stimulated peritoneal macrophages in vitro and in vivo in mice. 47 Interestingly, in a mouse model of renal I/R, relaxin was able to blunt the inflammatory response by reducing IL-1 and IL-18 levels, among other inflammatory markers. 48 For these reasons, contemplating that relaxin, possibly through NO production, could inhibit the NLRP3 inflammasome is plausible. Further studies are needed to address this hypothesis.
Extracellular Matrix
Multiple animal models have evaluated the role of relaxin in modifying extracellular matrix deposition and turnover in several organ systems, including the cardiovascular system. Relaxin treatment of cardiac fibroblasts decreased collagen types I and III, attenuated fibroblast differentiation and proliferation, and increased expression of matrix metalloproteinase (MMP) 2 in the fibroblasts subjected to angiotensin II and TGF-β. 49 The study took advantage of 2 models of fibrotic cardiomyopathy, one using relaxin-null mice and the other bearing cardiac-restricted overexpression of the β2-adrenergic receptor. Following continuous relaxin infusion for 14 days in the 2 strains of mice, collagen expression was reduced by 40% and 58%, respectively. Moreover, in a rat model of isoproterenol-induced cardiomyopathy, relaxin attenuated the increase in interstitial collagen accumulation and cardiac hypertrophy. 50 Relaxin also mitigated cardiac fibrosis in a mouse model of AMI. 51 In this study, 2 groups of mice were treated with continuous infusion of relaxin over 7 or 30 days after MI. In both groups, relaxin reduced TGF-β expression, myofibroblast differentiation, and increased MMP-13 levels. Despite these findings, relaxin did not negatively impact the incidence of ventricular free wall rupture or the extent of ventricular remodeling and dysfunction, indicating a favorable outcome that preserves reparative fibrosis.
Cardiac Regeneration
Cellular cardiomyoplasty involves myogenic cell grafting of dead muscle using autologous skeletal myoblasts as a method for regeneration of myocardial tissue after infarction. 52
In a rat model of ischemic HF, 53 combination therapy with retrograde coronary venous infusion of myoblasts and systemic relaxin resulted in improved cardiac function and myocardial viability when compared with myoblast cardiomyoplasty alone, systemic relaxin alone, or control. Additionally, histological analysis showed that myoblasts settled in ischemic scarring regions, especially with relaxin treatment. These results suggest that relaxin may have a synergistic effect with myoblasts in counteracting postischemic remodeling.
Relaxin: Clinical Approach to Cardiovascular Disease
Heart Failure
The well-established role of relaxin in cardiovascular and renal adaptation during pregnancy, together with the evidence in observational studies showing that elevated relaxin plasma levels correlated with the severity of HF patients, 10 suggested that relaxin may have beneficial effects in these patients.
The Pre-RELAX-AHF, 54 a phase 2, dose-finding study with 234 participants, suggested beneficial effects of serelaxin on both dyspnea and postdischarge clinical outcome in patients admitted for AHF with evidence of congestion, normal-to-raised blood pressure, and mild-to-moderate renal impairment. This led to the design of RELAX-AHF, 11 which was a phase III, prospective, randomized, double-blind, placebo-controlled, multicenter trial in the same targeted patient population that compared a 48-hour infusion of serelaxin versus placebo. Enrolled participants (n = 1161) were older than those enrolled in previous AHF studies, 55,56 with a mean age of 72 years, a mean SBP of 142 mm Hg, and nearly half the population had left ventricular ejection fraction greater than 40%.
Primary end points included change in dyspnea from baseline to day 5 quantified by the area under the curve (AUC) of a visual analog scale (VAS) score and moderate or markedly improved dyspnea at 6, 12, and 24 hours after drug initiation as measured by a 7-point Likert-type scale patient-reported dyspnea to the start of the study. Secondary end points were days alive and out of hospital for 60 days and cardiovascular death or readmission for HF or renal failure before 60 days. Serelaxin improved the VAS AUC dyspnea end point compared with placebo but had no significant effects on the other primary outcome or both secondary outcomes. Interestingly, serelaxin was associated with significant reductions in a prespecified additional end point, with fewer deaths at day 180 (placebo, 65 deaths; serelaxin, 42; hazard ratio [HR] 0.63, confidence interval [CI] 0.42-0.93; P = .019).
Dyspnea, however, is a subjective sensation, and currently there are no validated and reliable approaches to detect changes in dyspnea. 57 The fact that there was a significant benefit with serelaxin in only one of the dyspnea end points might be reflective of this notion. However, a recent hemodynamic study in patients with congestive HF showed that serelaxin exerted favorable hemodynamic effects on pulmonary capillary wedge pressure and mean pulmonary arterial pressure, 58 which could be consistent with dyspnea improvement. Although the reduction in postdischarge mortality at 180 days with serelaxin is very encouraging, some considerations need to be clarified. A limitation in this study is that it was not prospectively powered to assess mortality, and the mortality rate was 11% in the placebo group, which is at the lower end of the 10% to 20% range reported previously. 4 Nevertheless, a 48-hour drug infusion that may reduce mortality in AHF in the long term is certainly attractive and highly needed in this patient population. Several subanalyses of the RELAX-AHF were published in order to provide possible explanation of the 180-day survival benefit with serelaxin. 59 –61 No differences in outcomes were shown in the patient subgroup analysis. 60 In the 180-day, all-cause mortality end point, a larger reduction with serelaxin versus placebo was noted in patients aged ≥75 years (P = .0473), those with no HF hospitalization during the past year (P = .0222), blood lymphocytes ≤ 12% (P = .0298) and GFR <50 mL/min/m2 (P = .0286). 60 In another subanalysis, the reduction in the 180-day mortality with serelaxin was associated with fewer signs of organ damage measured by changes in cardiac, renal, and hepatic markers, and more rapid relief of congestion during the first days after admission. 61 These findings suggest that serelaxin may affect outcomes directly by organ protection. Another subanalysis assessed the effect of serelaxin on specific modes of death in patients with acute HF. 59 Here, serelaxin was shown to affect other cardiovascular deaths (HR: 0.29; 95% CI: 0.12-0.73; P = .005) and sudden death (HR: 0.46; 95% CI: 0.20-1.07; P = .065) but not death from HF (HR: 1.16; 95% CI: 0.61-2.21; P = .655). Although many mechanisms could explain the reason through which serelaxin may improve survival in patients with HF (anti-inflammatory, antifibrotic, and proangiogenic properties), the low numbers of deaths in a trial with limited statistical power narrows possible inferences on the impact of serelaxin on survival in patients with HF. To this end, the RELAX-AHF-2 trial was designed to address harder clinical outcomes, such as mortality as primary outcome, and target a larger population of 6375 patients (ClinicalTrial.gov Identifier: NCT01870778). All clinical trials to date investigating the potential therapeutic benefits of relaxin in cardiovascular disease are summarized in Table 1.
Summary of Clinical Trials Evaluating Relaxin in Cardiovascular Disease.
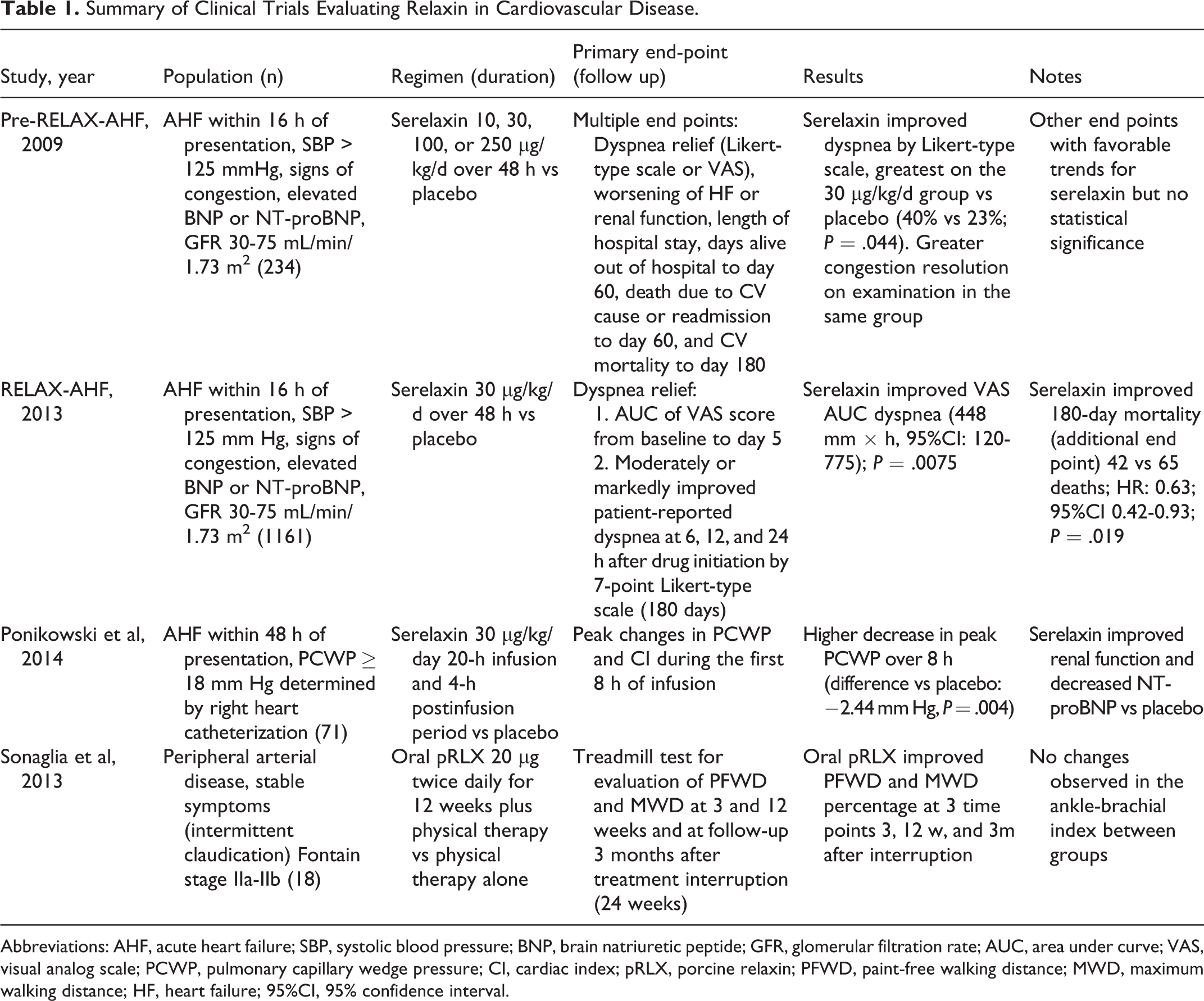
Abbreviations: AHF, acute heart failure; SBP, systolic blood pressure; BNP, brain natriuretic peptide; GFR, glomerular filtration rate; AUC, area under curve; VAS, visual analog scale; PCWP, pulmonary capillary wedge pressure; CI, cardiac index; pRLX, porcine relaxin; PFWD, paint-free walking distance; MWD, maximum walking distance; HF, heart failure; 95%CI, 95% confidence interval.
Acute Myocardial Infarction
Depletion of ATP during AMI leads to anaerobic metabolism, lactate accumulation, acidosis, and impairment of the ATPase sodium–potassium pump, 62 which culminates in loss of membrane integrity and cell death. The timely restoration of blood flow to the ischemic myocardium is not without limitation, as it can induce reperfusion injury and paradoxically increase infarct size. 63
Microvascular obstruction, or the “no-reflow” phenomenon, is a form of irreversible damage after reperfusion, which results in obstruction and disorganization of the capillary structure at risk due to tissue swelling, endothelial disruption, plugging by neutrophil accumulation, and microthrombi. 64 Lethal reperfusion injury, on the other hand, causes cardiomyocyte death from oxidative stress, calcium overload, rapid restoration of physiological pH, and inflammation. 62 Interestingly, relaxin seems to target many of these mechanisms of cell injury and has been shown to be protective in several models of I/R by reducing oxidative stress, calcium overload, neutrophil activity, endothelial adhesive protein expression, mast cell degranulation, apoptosis, platelet adhesion, as well as improving coronary blood flow, mostly via a mechanism involving NO. 12,13,33,38,39,65 Therefore, initiation of clinical trials to investigate the effects of relaxin in patients with AMI is enticing, although some considerations need to be addressed. Interventions in animal studies on myocardial I/R injury with promising “cardioprotective” drugs can reduce infarct size up to 50% 66 ; however, attempts to reproduce these effects in most clinical trials have been disappointing and raised many questions. Most of the I/R models that tested relaxin were ex vivo, and the drug was given before ischemia. 13,33 Moreover, although reperfusion therapy with relaxin in a swine model of I/R reduced infarct size, 12 long-term end points could not be established due to study design (including infarct size at 24 hours, survival analysis, and cardiac function with longer follow-up), altogether limiting clinical translational applicability. Therefore, future preclinical research is warranted to evaluate the effect of relaxin on long-term end points in the setting of AMI.
Peripheral Arterial Disease
Peripheral arterial disease is caused by chronic arterial occlusive disease of lower extremities due to atherosclerosis. According to Fontaine classification, symptoms vary from asymptomatic and intermittent claudication to ischemic leg pain at rest and trophic lesions. Given its properties in terms of vasodilation, platelet inhibition, and neoangiogenesis, the hormone relaxin could play a role in patients with PAD.
In an open-label pilot trial, 16 patients with PAD were randomly divided into 2 groups and treated with placebo or gastroprotected porcine oral relaxin for 3 months with concurrent physical therapy. 67 Patients had intermittent claudication as the main clinical manifestation of PAD and were corroborated with an arterial Echo-Doppler of the lower limbs. Primary end points were pain-free walking distance and maximum walking distance at 3 and 12 weeks of treatment and 12 weeks after treatment cessation. Interestingly, oral porcine relaxin improved these end points as early as 3 weeks from treatment initiation, and the benefits lasted throughout the 12 weeks after treatment cessation. 67 This study provides first clinical evidence of oral relaxin in long-term treatment in patients with PAD. Although this study has its limitations in terms of study design, the fact that oral relaxin could be a therapeutic option is interesting. This allows for long-term pharmacological treatment in other chronic cardiovascular diseases including chronic angina and chronic HF, since the current route of administration of serelaxin is via intravenous infusion. However, more studies are needed to carefully assess the plasma levels of relaxin following administration of oral porcine relaxin to confirm its role in the observed benefits in these patients.
Conclusion and Future Perspectives
Relaxin is a hormone with vasodilatory properties and pleiotropic effects in the cardiovascular system, which likely through a central mechanism involving NO has been shown to be cardioprotective in several animal models including I/R, hypertension, fibrotic cardiomyopathy, and MI. Despite all the mechanistic studies utilizing relaxin in preclinical models of cardiac disease, none of the proposed mechanisms explains the remarkable survival benefit in patients with AHF at 180 days, although the improvement in renal function and dyspnea may be attributed to NO signaling. Therefore, carefully designed preclinical mechanistic and translational studies with serelaxin are needed.
Translation into clinical practice, however, is a long and challenging route. The RELAX-AHF trial tested serelaxin in patients with AHF and showed reduction in overall mortality at 180 days. The RELAX-AHF-2 is currently ongoing, and it aims to evaluate the effects of serelaxin on survival in a larger population. Basic and preclinical research outcomes have indicated that relaxin may have the potential to attenuate AMI. Although translating these findings in patients with AMI is quite attractive, evaluating the safety profile of relaxin in the setting of emergent PCI and its effects in different animal models on survival, infarct size, inflammation, and remodeling prior to initiation of clinical trials is warranted. Finally, the development of oral porcine relaxin for treatment of PAD represents the first clinical study with oral delivery of relaxin and supports a potential new application for chronic treatment of patients with HF and stable coronary artery disease.
Footnotes
Authorship Contribution
Valle Raleigh, J contributed to conception and design, contributed to interpretation, drafted manuscript, gave final approval, agrees to be accountable for all aspects of work ensuring integrity and accuracy; Toldo, S contributed to design, contributed to interpretation, drafted manuscript, gave final approval, agrees to be accountable for all aspects of work ensuring integrity and accuracy; Das, A contributed to interpretation, critically revised manuscript, gave final approval, agrees to be accountable for all aspects of work ensuring integrity and accuracy; Abbate, A contributed to design, contributed to interpretation, critically revised manuscript, gave final approval, agrees to be accountable for all aspects of work ensuring integrity and accuracy; Salloum, F contributed to conception and design, contributed to interpretation, drafted manuscript, critically revised manuscript, gave final approval, agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Drs. Salloum and Abbate have received research grants from Novartis Pharmaceuticals.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Salloum is supported by a Grant-in-Aid from the American Heart Association (14GRNT20010003) and Investigator-Initiated Trials from Novartis Pharmaceuticals.