
Abstract
Lethal myocardial ischemia–reperfusion (I/R) injury has been attributed in part to mitochondrial respiratory dysfunction (including damage to complex I) and the resultant excessive production of reactive oxygen species. Recent evidence has shown that reduced nicotinamide adenine dinucleotide–quinone internal oxidoreductase (Ndi1; the single-subunit protein that in yeast serves the analogous function as complex I), transduced by addition of the TAT-conjugated protein to culture media and perfusion buffer, can preserve mitochondrial function and attenuate I/R injury in neonatal rat cardiomyocytes and Langendorff-perfused rat hearts. However, this novel metabolic strategy to salvage ischemic-reperfused myocardium has not been tested in vivo. In this study, TAT-conjugated Ndi1 and placebo-control protein were synthesized using a cell-free system. Mitochondrial uptake and functionality of TAT-Ndi1 were demonstrated in mitochondrial preparations from rat hearts after intraperitoneal administration of the protein. Rats were randomized to receive either TAT-Ndi1 or placebo protein, and 2 hours later all animals underwent 45-minute coronary artery occlusion followed by 2 hours of reperfusion. Infarct size was delineated by tetrazolium staining and normalized to the volume of at-risk myocardium, with all analysis conducted in a blinded manner. Risk region was comparable in the 2 cohorts. Preischemic administration of TAT-Ndi1 was profoundly cardioprotective. These results demonstrate that it is possible to target therapeutic proteins to the mitochondrial matrix and that yeast Ndi1 can substitute for complex I to ameliorate I/R injury in the heart. Moreover, these data suggest that cell-permeable delivery of mitochondrial proteins may provide a novel molecular strategy to treat mitochondrial dysfunction in patients.
Keywords
Introduction
Myocardial ischemia–reperfusion (I/R) injury is attributed in part to mitochondrial damage and dysfunction, including I/R-induced defects in complex I activity. 1 Complex I (reduced nicotinamide adenine dinucleotide [NADH]-quinone oxidoreductase) is the first component of the electron transport chain that catalyzes the transfer of electrons from NADH to ubiquinone. It is composed of 45 subunits and resides in the inner mitochondrial membrane. This enzyme complex plays a critical role in oxidative phosphorylation (OXPHOS) and contributes to the formation of the proton gradient that drives adenosine triphosphate (ATP) synthesis.
There is considerable evidence that complex I is a major site of damage during I/R; its activity rapidly falls during both ischemia and reperfusion. 2 –7 The decline in complex I function may be even greater during reperfusion. 1 The defect in complex I activity in the I/R-injured rat heart has been linked to oxygen free radical-induced cardiolipin damage. Complex I 1 and complex III 8 are known sources of reactive oxygen species (ROS) and require cardiolipin in the inner mitochondrial membrane for functionality. Thus, in the setting of I/R injury, a vicious cycle is established in which ROS from complex I damages cardiolipin, which impairs normal electron flow through complex I and increases ROS production. Thus, complex I represents a potential therapeutic target for amelioration of I/R injury.
One approach to restore normal NADH oxidation and electron flow at reperfusion is xenotransplantation of NADH-quinone internal oxidoreductase (Ndi1). 9 Reduced nicotinamide adenine dinucleotide–quinone internal oxidoreductase is a single 56 kDa polypeptide that serves as the entry site of electrons from NADH to the respiratory chain in yeast and bacteria. Structurally simpler than the multi-subunit complex I of mammalian mitochondria, Ndi1 is functionally analogous with respect to oxidation of NADH and reduction of quinone; however, it does not pump protons. Heterologous expression of this yeast polypeptide in complex I-deficient mammalian cells partially restores NADH oxidation and ATP production, which can proceed via protons pumped from complexes III and IV.
Reduced nicotinamide adenine dinucleotide–quinone internal oxidoreductase was first studied by Seo and colleagues 10 as a possible treatment for Parkinson disease, based on the rationale that excess ROS from complex I contributed to the death of dopaminergic neurons in the substantia nigra. 11 –13 Neural cells transduced with virally delivered Ndi1 exhibited reduced levels of ROS production after rotenone poisoning. Utilization of NADH by Ndi1 was suggested to limit the availability of electrons to complex I for ROS production. 13 Based on these findings, we hypothesized that Ndi1 could attenuate ROS production from complex I damaged by myocardial I/R. To overcome the limitations of gene therapy, we generated a cell-permeable fusion protein of Ndi1 and the 11aa transduction domain from HIV-TAT (TAT-Ndi1) and demonstrated that it entered cardiomyocytes and localized to the mitochondrial inner membrane. 9 When the fusion protein was delivered to Langendorff-perfused rat hearts subjected to I/R, ROS production was suppressed, ATP levels were preserved, and infarct size was reduced by 62%. Similar production was observed when TAT-Ndi1 was infused at the onset of reperfusion. 9 We concluded that Ndi1 could reduce I/R injury in buffer-perfused hearts. The purpose of the present study was to establish whether Ndi1 could be used to functionally replace complex I and reduce myocardial infarct size in vivo.
Methods
Reduced Nicotinamide Adenine Dinucleotide–Quinone Internal Oxidoreductase Synthesis
TAT-conjugated Ndi1 protein was synthesized from a hemagglutinin (HA)- and histidine-tagged TAT-Ndi1 construct (provided by R. Gottlieb; prepared as described 9 ) using a cell-free protein synthesis kit (EasyXpress; Qiagen, Germantown, Maryland) according to the manufacturer’s instructions. This Escherichia coli lysate system resulted in higher yields and more full-length protein than bacterial expression (Figure 1). After low-speed centrifugation to remove debris, the protein product was concentrated using a 50 kDa cutoff, diluted with purification buffer (pH∼7.8, 50 mmol/L Tris, 200 mmol/L NaCl), and the fraction containing full-length Ndi1 protein was separated by high-performance liquid chromatography. Synthesis of tagged Ndi1 protein (molecular weight ∼75 kDa) was confirmed by immunoblotting for HA and Ndi1 (gift from the Yagi laboratory; Figure 1). Control DNA was used with the same Qiagen cell-free system to generate the material for control injections. Protein was stored at −80°C until use (<1 month).

Preparation of TAT-Ndi1. Immunoblot of TAT-Ndi1 purified from cell-free expression system. Lane 1, preparation using control DNA; lanes 2 to 5, preparation using cDNA for TAT-Ndi1; lane 6, lysate from cell-free expression system; lane 7, E. coli bacterial expression of TAT-Ndi1. Upper panel is probed with anti-HA antibody; lower panel is probed with anti-Ndi1 antibody. Arrowheads indicate 75 kDa, the expected MW of TAT-Ndi1. cDNA indicates complementary DNA; E. coli, Escherichia coli; HA, hemagglutinin; MW, molecular weight; Ndi1, nicotinamide adenine dinucleotide–quinone internal oxidoreductase.
Administration of TAT-Ndi1
The animal studies were approved by the Institutional Animal Care and Use Committee of Wayne State University and performed in accordance with the Guide for the Care and Use of Laboratory Animals. Female Sprague-Dawley rats received TAT-Ndi1 or placebo (2 mg protein/kg in a volume of 0.5 mL via intraperitoneal [ip] injection). The dose was established on the basis of preliminary experiments in which TAT-Ndi1 was given at doses ranging from 1 to 4 mg protein/kg; the presence of tagged TAT-Ndi1 in the heart (detected by immunoblotting) and reduction of infarct size (compared to historical controls) served as the primary end points (data not shown). Two hours after ip injection, rats were subjected to myocardial I/R as described subsequently. After 2 hours of reperfusion, hearts were harvested for determination of infarct size or for complex I assays.
Mitochondrial Isolation
Female Sprague-Dawley rats (weight: 368 ± 28 g) were randomized to receive TAT-Ndi1 (n = 4) or placebo (n = 3), followed 2 hours later by coronary artery occlusion. Two rats developed lethal ventricular fibrillation during the initial 20 minutes of ischemia. In the remaining 5 animals (2 TAT-Ndi1-treated rats, 3 placebo controls), the anterior left ventricular (LV) wall was harvested for analysis at 1-hour postperfusion. Two additional rats were used as untreated and nonischemic controls, with samples obtained under anesthesia in a time-matched manner from the anterior LV wall. Heart tissue was immersed in ice-cold mitochondrial isolation buffer (pH 7.4; mmol/L: sucrose 250, Tris-HCl 50, sodium hydroxyethyl piperazineethanesulfonic acid 10, EDTA 5, ethylene glycol tetra acetic acid 10) with a protease inhibitor cocktail and minced with scissors. The tissue suspension was transferred to an isolation buffer (pH 7.4) supplemented with bovine serum albumin (BSA; 0.5%), homogenized with 3 short pulses, and centrifuged at 1000g for 10 minutes at 4°C to remove debris. The supernatant was collected and centrifuged at 10 000g for 15 minutes at 4°C. The pellet was resuspended and recentrifuged, and the final pellet was resuspended in the isolation buffer. Protein concentration in the crude mitochondrial fraction was determined by bicinchoninic acid assay.
Mitochondrial Complex I Activity Determination
Mitochondrial complex I activity (ie, oxidation of NADH to nicotinamide adenine dinucleotide [NAD+]) was quantified using an adaptation of spectrophotometric methods. 14 –16 We compared total NADH oxidoreductase activity to rotenone-insensitive activity (attributable to Ndi1). 17,18 Aliquots of crude mitochondrial fraction (10 µL, containing 20 µg protein) were added to wells containing 120 µL assay buffer (25 mmol/L K2HPO4, 5 mmol/L MgCl2, 0.25% BSA, 3.7 µmol/L antimycin A, 2 mmol/L KCN, 62.8 mmol/L ubiquinone, pH 7.4). After 1 minute at 30°C, reactions were initiated by the addition of NADH (5 µL, 5.7 mmol/L) in the presence or absence of rotenone, a complex I inhibitor (2 µL, 0.36 mmol/L in dimethyl sulfoxide). The decrease in absorbance at 340 nm (oxidation of NADH) was monitored for 10 minutes. For all samples (±rotenone), the absorbance change reached a plateau within 3 minutes. The rate of NADH oxidation over this 3-minute interval was reported as absorbance units per minutes per milligram protein for total activity and rotenone-resistant activity (mean ± standard error).
Coronary Artery Occlusion and Infarct Size Determination
Adult female Sprague-Dawley rats (n = 21) weighing 327 ± 62 g (mean ± SD) were randomized to receive either TAT-Ndi1 or placebo-control protein (2 mg protein/kg in a volume of 0.5 mL ip). Ninety minutes after ip administration of protein, rats were anesthetized with sodium pentobarbital (50 mg/kg ip), intubated via tracheostomy, and ventilated with air. The heart was exposed through a left lateral thoracotomy at the fourth intercostal space, and the proximal left coronary artery was snare occluded for 45 minutes (initiated exactly 2 hours after ip administration of protein) followed by 2 hours’ reperfusion as described previously. 19,20 Hearts were harvested for mitochondrial isolation (using the anterior LV wall, as described previously) or for infarct size determination. The area at risk (AR) of infarction (the primary determinant of infarct size in this model) and the area of necrosis (AN) were assessed using standard methods. 20 –24 Briefly, AR was delineated by ligating the left coronary artery and injecting Monastral blue pigment into the vasculature via a cannula inserted into the femoral vein. Rats were then euthanized under deep pentobarbital anesthesia by injection of KCl. The hearts were rapidly excised, cut into 5 to 6 transverse slices, and photographed. To distinguish necrotic from viable myocardium, the heart slices were incubated in triphenyltetrazolium chloride for 10 minutes at 37°C and rephotographed. Both AR and AN were quantified using image analysis software (SigmaScan Pro; SPSS Inc, San Jose, California), corrected for tissue weight, and summed for each heart. Risk regions and infarct sizes were quantified by a single investigator who was blinded with regard to the group assignments. Area at risk was expressed as a percentage of the total LV weight, and rats in either group with a negligible risk region (prospectively defined as <10% of the LV 22 ) were excluded from further analysis. For remaining animals, AN was calculated and expressed as a percentage of the AR. For the 21 rats studied, 1 died at the time of anesthesia, 3 were excluded because of technical failures, and an additional 2 rats were excluded because of small risk regions occupying <10% of the LV. Data are reported for the 15 animals that successfully completed the protocol: 8 treated with TAT-Ndi1 and 7 placebo controls. Results are reported as mean ± standard error, and AR/LV and AN/AR were compared between TAT-Ndi1- and placebo-treated groups by t test.
Results
TAT-Ndi1 Transduction and Complex I Activity
Western blot analysis of heart mitochondria confirmed the presence of TAT-Ndi1 in heart mitochondria from rats that received TAT-Ndi1 by ip injection (Figure 2). Complex I activity (NADH oxidation) in nonischemic shams, assessed in the absence of rotenone, averaged 1.28 ± 0.04 U/min/mg. As expected, addition of rotenone inhibited complex I activity by 90% ± 5% to a mean of 0.13 U/min/mg (Figure 2). In all rats subjected to I/R, complex I activity in the absence of rotenone was attenuated when compared with shams, averaging 0.72 and 0.86 U/min/mg in the placebo controls and TAT-Ndi1-treated groups. In the placebo-control cohort, addition of rotenone inhibited complex I activity to the same degree as seen in the shams, that is, by 85% ± 5%, to a mean of 0.13 U/min/mg. In contrast, in TAT-Ndi1-treated rats, complex I activity was only partially inhibited by rotenone to a mean of 0.44 U/min/mg (Figure 2). Rotenone-insensitive NADH oxidase activity in cardiac mitochondria from TAT-Ndi1-treated rats reflects the contribution of Ndi1. Because of the limited number of samples obtained, statistical analysis of the 3 groups was not done.
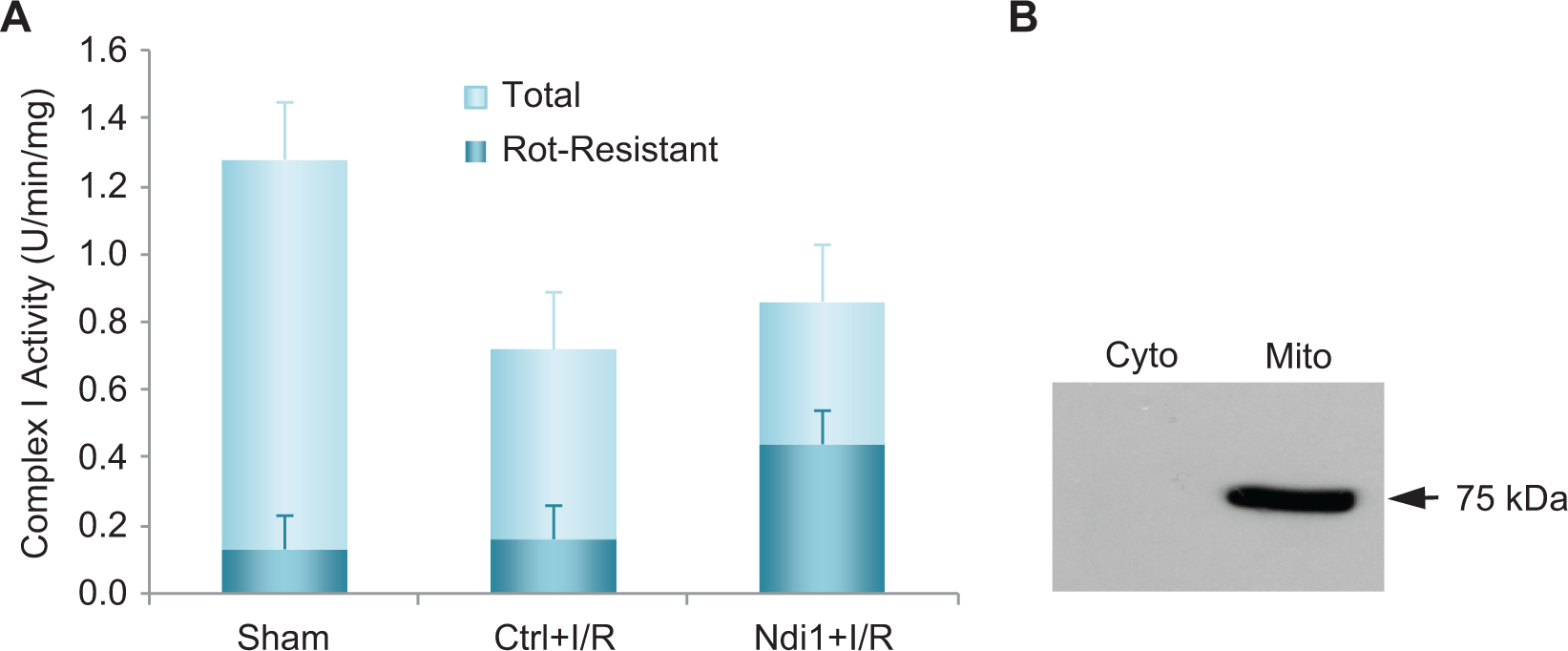
Complex I and Ndi1 activity in rat heart mitochondria. A, total and rotenone-resistant complex I activity was measured in rat heart mitochondria from control rats (n = 2); placebo protein-treated rats subjected to I/R (ctrl + I/R; n = 3); and TAT-Ndi1-treated rats subjected to I/R (Ndi1 + I/R; n = 2). B, Blot of cytosol and mitochondria from rat heart 2 hours after ip administration of TAT-Ndi1 probed with anti-HA antibody. Statistical analysis was not done due to the limited number of samples obtained. Ctrl indicates control; ip, intraperitoneal; I/R, ischemia–reperfusion; HA, hemagglutinin; Ndi1, nicotinamide adenine dinucleotide–quinone internal oxidoreductase.
Effect of TAT-Ndi1 on Infarct Size
To determine whether pretreatment with TAT-Ndi1 could ameliorate I/R injury, we administered TAT-Ndi1 or placebo 2 hours before I/R. The 2 groups were well matched with regard to risk region: AR/LV averaged 32% ± 5% and 29% ± 3% in control and TAT-Ndi1-treated rats, respectively (P = .800, not significant). However, infarct size was significantly smaller in rats that received TAT-Ndi1 versus placebo controls (33% ± 6% vs 60% ± 5%; P = .005; Figure 3). Thus, ip administration of TAT-Ndi1 was able to protect cardiac mitochondria from I/R injury and reduce infarct size in an in vivo model.

Effect of TAT-Ndi1 on infarct size. Infarct size as a percentage of the risk region (28%-32% of total LV weight: no difference between groups). Solid circles represent animals treated with control protein, and shaded squares represent animals treated with TAT-Ndi1. Open symbols indicate mean ± SE; P = .005. Representative TTC-stained heart sections are shown from rats treated with control protein or TAT-Ndi1. LV indicates left ventricular; TTC, triphenyltetrazolium chloride; SE, standard error; Ndi1, nicotinamide adenine dinucleotide–quinone internal oxidoreductase.
Discussion
We provide novel evidence that TAT-Ndi1 reduced infarct size in an in vivo model of I/R injury. Administration of Ndi1, the yeast polypeptide equivalent of mammalian complex I, contributed to the NADH oxidase activity of isolated cardiac mitochondria. Immunoblotting for TAT-Ndi1 and the presence of rotenone-insensitive NADH oxidase activity indicate that enzymatically active protein was present in the mitochondria.
Our studies confirm reports by others that complex I activity is reduced after I/R. 25 –32 Our measurement of complex I activity did not show an increase in activity in the cardiac mitochondria from animals treated with TAT-Ndi1, in contrast to our previous findings in the ex vivo model. This may be related to the assay method (respirometry in the Perry study 9 and isolated NADH oxidase activity in the present work). In both studies, however, the contribution of Ndi1 to overall complex I activity was modest. The previous findings suggested that TAT-Ndi1 protected the heart by accelerating NADH oxidation, thus decreasing the availability of these electrons for ROS production by damaged complex I and other NAD(P)H oxidases. Thus, TAT-Ndi1 may reduce I/R injury by supporting electron flow through the respiratory chain, and perhaps more importantly by preventing ROS production from excess NADH. The mechanism of TAT-Ndi1 cardioprotection was previously addressed, 9 and therefore it was not re-examined in the study. The primary purpose of this study was to demonstrate cardioprotective efficacy in vivo.
Another approach to cardioprotection involving NADH-ubiquinone oxidoreductase is the reduction of ROS production by endogenously inhibiting complex I. Using mitochondria-targeted nitrosothiols (MitoSNOs), Prime et al 33 reported that infusion of the compound MitoSNO1 mimicked ischemic preconditioning and was protective against I/R injury in Langendorff-perfused mouse hearts when administered at reperfusion. Hearts had better recovery of function as assessed by rate pressure product and a decrease in infarct size. They surmised that the protection conferred by MitoSNO1 was likely a consequence of the persistent S-nitrosylation of complex I. Subsequently, Chouchani et al 34 directly showed that MitoSNO S-nitrosylation of cysteine 39 of the ND3 subunit of complex I was associated with reversible inhibition of the activity of complex I during ischemia, with benefits during the early minutes of reperfusion. Their studies indicated that the S-nitrosylation interfered with electron transfer to ubiquinone but not the interaction with NADH at the flavin center. Interestingly, S-nitrosylation of ND3 does not result in ROS production, in contrast to rotenone, a complex I inhibitor, which binds to the ND1 subunit (not to be confused with yeast Ndi1). This may be due to differential effects on retrograde electron flow (complex II → ubiquinone → complex I), which conceivably may be prevented by S-nitrosylation of ND3 but not by rotenone. NADH-quinone internal oxidoreductase, by providing an alternative path for electron flow to ubiquinone that does not involve a reactive semiquinone intermediate, protects the mitochondria during reperfusion while supporting ATP production and NADH oxidation. Although Chouchani et al 34 did not measure ATP or NADH/NAD+ ratio, their findings suggest that S-nitrosylating agents could be beneficial in the setting of I/R injury. It is possible that concurrent administration of TAT-Ndi1 and MitoSNO could have additive benefit, assuming MitoSNO does not inhibit Ndi1. NADH-quinone internal oxidoreductase supports electron transfer from NADH to ubiquinone to support OXPHOS, whereas S-nitrosylation of ND3 would mitigate ROS production from endogenous complex I.
The novel finding in our study is the demonstration for the first time that it is possible to deliver a cell-permeable functional enzyme to the mitochondrial matrix in vivo. TAT-Ndi1 persists in heart tissue for at least 48 hours, when administered ip to mice (see Supplemental data), so its beneficial effects might persist long enough to permit replacement of endogenous complex I damaged by I/R injury. It may also be possible to treat inheritable mitochondrial diseases such as complex I deficiencies with therapeutic protein delivery. TAT-Ndi1 could offer benefit in the setting of acute poisoning with rotenone or the parkinsonian mimetic 1-methyl-4-phenylpyridinium. The administration of a yeast protein may theoretically elicit an immunologic response that could limit its use for chronic therapy, but long-term expression of Ndi1 (mediated by viral gene transfer) did not elicit an immune response, whereas cytosolic expression of green fluorescent protein did. They attributed the lack of immune response to Ndi1 to its intramitochondrial localization. 35 It remains to be determined whether TAT-Ndi1 will elicit an immune response if given repeatedly, which could restrict its use to one-time administration.
TAT-Ndi1—even as a one-time intervention—could be useful in the setting of acute myocardial infarction. Perry et al showed that TAT-Ndi1 could reduce infarct size when administered at reperfusion, 9 although the present study did not examine infusion of the protein at reperfusion. TAT-Ndi1 could also improve organ preservation for transplantation and might enhance bridge to recovery after placement of a LV-assist device.
In summary, this study demonstrates feasibility and efficacy of therapeutic protein administration for mitochondrial dysfunction.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported in part by NIH R01 HL034579 to RMM and RAG and NIH R01 HL060590 to RAG.