
Abstract
The dawn of cardioprotection by infarct size reduction originated from the idea to favourably alter the oxygen demand–supply balance of the ischaemic/infarcting myocardium by reducing the contractile determinants of its oxygen consumption. This idea is probably not correct, since the ischaemic/infarcting myocardium does not contract anyway. None of the successful initial preclinical attempts of infarct size reduction translated into clinical practice, except for timely reperfusion which has become and still is the backbone of all clinical infarct therapy up today. The idea of cardioprotection gained momentum again with the recognition of ischaemic conditioning, and a myriad of preclinical studies have identified molecules and mechanisms of such self-defence mechanism. Although there are positive clinical proof-of-concept studies, ischaemic conditioning strategies and drugs related to its signal transduction have not translated into clinical practice. We are currently trying to understand the obstacles to translation from successful preclinical studies on cardioprotection to clinical practice, but are also waiting for an innovative mechanistic breakthrough.
First Wave of Cardioprotection Studies, but Persistent Success and Translation Only for Timely Reperfusion
The modern era in the treatment of acute myocardial infarction, beyond that traditional one by bed rest and symptomatic treatment of arrhythmias and hemodynamic complications, began in the late sixties/early seventies of the last century (Figure 1). Stimulus and backbone for these new treatment approaches was Braunwald's revival of Rein's and Büchner's original ideas of ‘coronary insufficiency’ as an imbalance of oxygen supply and demand and his extension of this paradigm to acute myocardial infarction and its treatment.1,2 Braunwald and his colleagues had previously elaborated in global dog heart preparations on the hemodynamic determinants of myocardial oxygen consumption3,4 and now used treatment approaches that attenuated the supply–demand imbalance by reducing these hemodynamic determinants of myocardial oxygen consumption, such as beta blockers or calcium antagonists. 5 Propranolol indeed reduced heart rate and infarct size (as measured by myocardial creatine phosphokinase concentration) in an anaesthetised open-chest dog preparation with coronary occlusion. 5 These experiments were extended to enhance myocardial metabolism by glucose-insulin-potassium, 6 to hyaluronidase to increase the permeation of metabolites from the vasculature into cardiomyocytes and attenuate edema7,8 and to corticosteroids to attenuate inflammation. 9 The enthusiasm over these novel approaches to treat acute myocardial infarction did not translate well into clinical practice. An initial randomised clinical trial on propranolol versus standard treatment in patients with acute myocardial infarction failed to demonstrate a reduction in infarct size. 10 The reduction of infarct size by beta blockade is equivocal up today, even for the ß1-selective blocker metoprolol,11,12 notwithstanding the benefits of beta blockade for arrhythmias and postinfarct remodeling. The use of glucose–insulin–potassium in acute myocardial infarction is also equivocal up today. 13 For the use of corticosteroids in acute myocardial infarction, concern was raised for the potential to impair scar formation and facilitate myocardial rupture.14,15 However, this concern never materialised. 16 Given subsequent experimental evidence for the benefit of corticosteroids in coronary microembolisation17,18 and the more recent evidence for benefit from colchicine in patients with acute myocardial infarction, 19 I wonder whether it may have been premature to not pursue corticosteroids. From my personal perspective, the oxygen supply–demand imbalance paradigm, however, was and is not correct for use in acute myocardial infarction, since the ischaemic/infarcting myocardium does not contract anyway and thus a reduction of the contractile determinants of myocardial oxygen consumption has little impact. In fact, the ischaemic and infarcting myocardium is entirely dependent on oxygen supply by coronary blood flow through collaterals and can therefore only to some extent benefit from a redistribution of blood flow towards the ischaemic myocardium after reduction of contractile function, metabolism and blood flow in the non-ischaemic myocardium. 20 Therefore, beta blockade and notably the decreased heart rate increase blood flow to the ischaemic myocardium at the expense of non-ischaemic myocardium and, in fact, increase its contractile function and presumably oxygen consumption.21–23
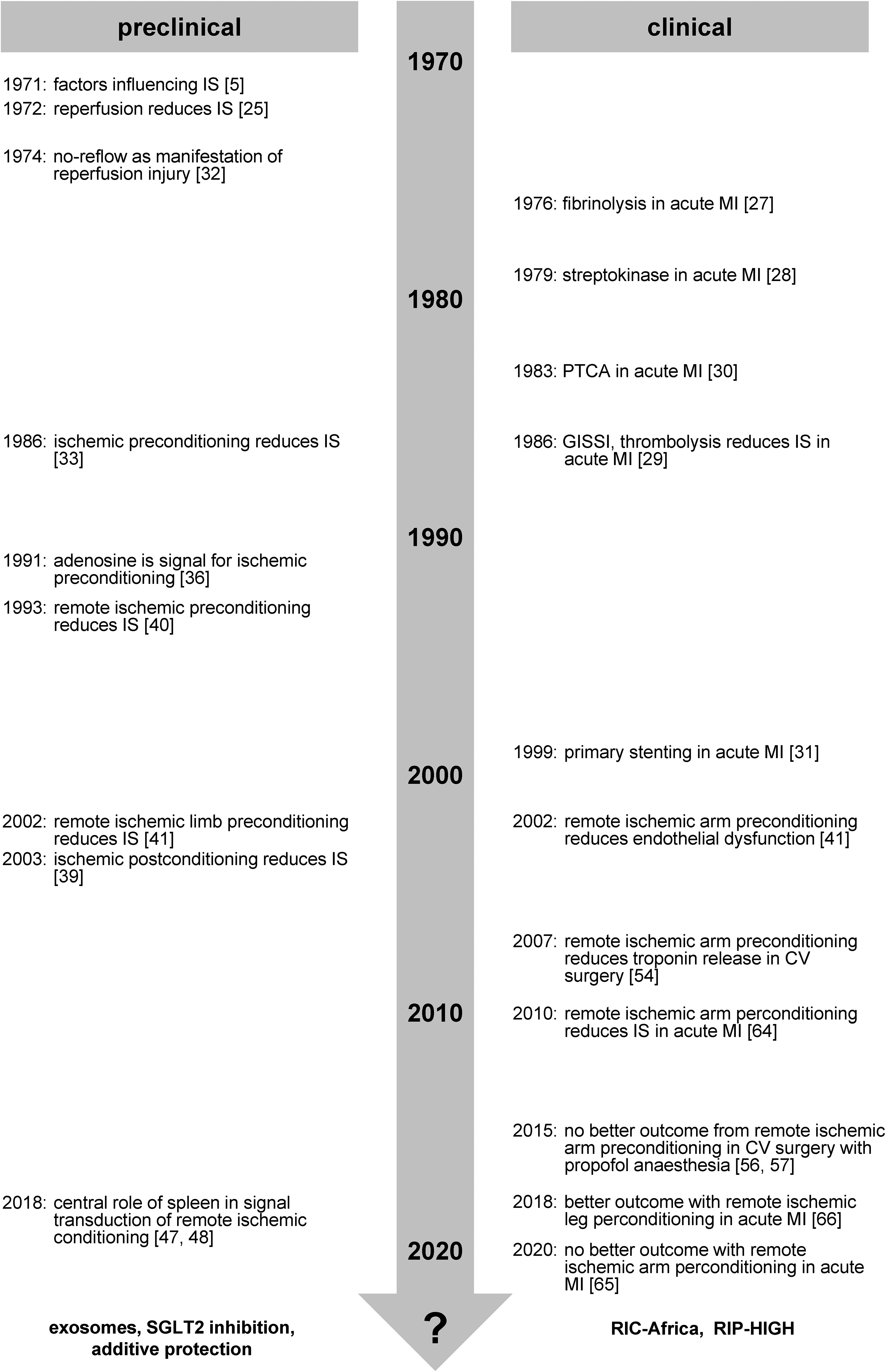
Time line of landmark events in preclinical and clinical cardioprotection research. CV, cardiovascular; IS, infarct size; MI, myocardial infarction.
The real breakthrough of the modern era in the treatment of myocardial infarction which was spearheaded by Ross was the recognition that reperfusion after 3 h coronary occlusion reduced infarct size in a dog preparation.24,25 Already a few years later, as detailed elsewhere, 26 Chazov et al 27 and briefly thereafter Rentrop et al 28 used thrombolysis to induce reperfusion and treat patients with acute myocardial infarction. The GISSI trial then provided definitive proof for the benefits of reperfusion by thrombolysis in patients with acute myocardial infarction. 29 In parallel, mechanical reperfusion by percutaneous transluminal coronary angioplasty 30 and subsequently stenting 31 was developed. Undoubtedly, primary percutaneous coronary intervention (primary PCI) is today the gold standard therapy of acute myocardial infarction. Accordingly, any cardioprotective intervention will have to prove its benefit beyond that by rapid reperfusion. However, only 2 years after the seminal recognition of reperfusion as the prime strategy to reduce infarct size,24,25 the no-reflow phenomenon was identified as a manifestation of reperfusion injury and inevitable downside of reperfusion. 32
Second Wave of Cardioprotection Studies – Ischaemic Conditioning, its Signal Transduction and Related Therapy
A novel paradigm of cardioprotection evolved with the recognition of ischaemic preconditioning by Murry et al, 33 a cardioprotective intervention which is only effective in conjunction with timely reperfusion. Apparently, one or several brief cycle(s) of coronary occlusion/reperfusion preceding a more sustained period of coronary occlusion which causes infarction can initiate the activation of a molecular self-defence program which delays the temporal and spatial progression of infarction such that, with timely reperfusion, the resulting infarct size is reduced.34,35 The recognition of ischaemic preconditioning gained more momentum when Downey and colleagues identified the formation and activation of signalling molecules, eg adenosine 36 and protein kinase C, 37 which were causal for the infarct size reduction by ischaemic preconditioning. Since then, a myriad of signalling steps, including calcium, nitric oxide, reactive oxygen species, autacoids, hormones, neurotransmitters, and cytokines which activate ion channels and intracellular enzymes in various subcellular compartments to ultimately delay various modes of cell death have been identified in various species, models and preparations and related to the efficacy of ischaemic conditioning.35,38 The original preconditioning paradigm has subsequently been extended to the activation of a self-defence program also following (postconditioning) 39 coronary occlusion/reperfusion. Also, such self-defence program can be initiated at a distance (remote ischaemic conditioning),40,41 and this form of protection can even be recruited during ongoing coronary occlusion. 42 The study of the signal transduction of all these ischaemic conditioning maneuvers has been very fruitful and has identified numerous protective molecules and mechanisms, including novel modes of cell death, regulated and non-regulated. 43 Interestingly, not all modes of cell death contribute to infarction, and autophagy and its initiation may even be cardioprotective.44,45 Cardioprotection by remote ischaemic conditioning turned out to be a manifestation of a more generalised organ-to-organ adaptive and protective response which involves neuronal and humoral pathways 46 and, of note, the spleen as neuro-humoral interface/relay organ.47,48
While basic research on ischaemic conditioning and its signalling has generated a wealth of fundamental novel knowledge, the translation of ischaemic conditioning interventions and of drugs related to ischaemic conditioning's signalling to the benefit of patients with acute myocardial infarction has been largely disappointing.49,50 Due to the unpredictable occurrence of myocardial infarction, ischaemic preconditioning is not feasible; ischaemic preconditioning can, however, be used in cardiovascular surgery, has reduced markers of injury but did not really provide benefit beyond that by cardioplegic arrest. 49 Ischaemic postconditioning reduced creatine kinase release in a smaller proof-of-concept trial in patients with acute ST segment elevation infarction 51 but not in the more recent phase III DANAMI 3 i-POST trial, 52 and the protective effects of ischaemic postconditioning in patients with acute myocardial infarction remain equivocal.35,49 The most attractive ischaemic conditioning intervention to be used in patients is certainly remote ischaemic conditioning 53 which is non-invasive and safe when using brief cycles of limb ischaemia/reperfusion by inflation/deflation of a blood pressure cuff. 41 However, whereas initial smaller proof-of-concept trials in patients undergoing cardiovascular surgery54,55 reported reduction of troponin release, the following larger phase III trials were neutral, with respect to troponin release and clinical outcome in patients undergoing cardiovascular surgery.56,57 In cardiovascular surgery, anaesthesia per se, notably volatile anaesthesia, provides some cardioprotection, which can possibly be even augmented with use of noble gases such as argon or xenon.58,59 Remote ischaemic conditioning is operative in patients undergoing cardiovascular surgery during isoflurane anaesthesia but protection is abrogated during propofol anaesthesia,60,61 thus possibly explaining the neutral results of the larger trials which had mostly used propofol. 62 Sodium-proton exchange inhibition in patients undergoing surgical coronary revascularisation reduced the incidence of myocardial infarction but nevertheless increased mortality, associated with increased cerebrovascular events, over 5 days. 63 In patients with ST segment elevation infarction undergoing PCI an initial trial (CONDI 1) reported myocardial salvage, 64 but the subsequent trials were equivocal – the larger ERIC-PPCI/CONDI 2 trial was neutral with remote ischaemic conditioning at the upper limb, 65 but the smaller RIC-STEMI trial with remote ischaemic conditioning at the lower limb demonstrated reduced mortality and hospitalisation for heart failure. 66 Pharmacological approaches using substances related to the signal transduction of ischaemic conditioning, including eg adenosine, cyclosporine, PKC delta inhibition, have also – after preliminary more promising data – not resulted in robust infarct size reduction and improved clinical outcome in patients with reperfused acute myocardial infarction.35,50 Also, other, non-pharmacological approaches which had resulted in promising experimental data on infarct size reduction have not been successfully translated to patient benefit, including gentle reperfusion, 67 mild hypothermia,68,69 and hyperoxia.70–72
Waiting for a Third Wave of Cardioprotection – Understanding the Obstacles
Potential reasons for the translational gap between preclinical and clinical studies on cardioprotection are multifold and have been discussed extensively. Certainly, this is not simply a disconnect between humans and other species. From my personal perspective, reasons for the translational gaps are present on both the preclinical and the clinical side. Preclinical studies are reductionist and mechanistic by their very nature. Therefore, most studies in various experimental preparations and models in various species have selected an ischaemic conditioning protocol which best worked in the specific laboratory to then identify signalling molecules and mechanisms; when using animals, such studies were mostly done in young and healthy animals and in the presence of anaesthesia. Preparations, models and protocols which did not result in infarct size reduction were discarded and most often not reported. Only recently, with the recognition of the translational gap, have studies in rats73,74 and pigs75,76 reported failure of infarct size reduction by ischaemic conditioning. 77 Primordial non-responsiveness to cardioprotection by a given intervention, which is possibly determined by the genome, has been neglected so far and may account for some of the translational gap. Then, a given cardioprotective intervention and protocol may work in one laboratory, but not be robust against the intervening variables of animal species and strain, age, sex, anaesthesia, adjunct medications (heparin, platelet inhibitors, anti-arrhythmics) and differences in dose, timing and algorithm of the cardioprotective intervention and therefore not work in another laboratory, and even less in patients where intervening variables also differ between different clinics. Also, such lack of robustness has been neglected – in part, because publication of such studies was considered as not innovative and mechanistic – and has only recently been put forward.77,78 The best investigated obstacle to translation on the preclinical side is the lack of co-morbidities (hypertension, diabetes, hypercholesterolemia, etc.) and co-medications (statins, platelet inhibitors, anti-diabetics, etc.) which patients with acute myocardial infarction typically have, and which interfere with cardioprotection79,80; some of these co-medications induce cardioprotection per se (eg, morphine, P2Y12 inhibitors) and thus reduce the potential for further cardioprotection by the intervention under study, whereas others impair cardioprotective signalling. There is solid preclinical evidence for impairment of cardioprotective signalling by comorbidities and comedications, but the clinical evidence is much less solid. 79 The clinical obstacles to translation of cardioprotection to the benefit of patients with myocardial infarction mostly relate to patient selection. There is a time window for cardioprotection to be effective – with a short duration from symptom onset to reperfusion no adjunct protection is needed, whereas with a too long duration of coronary occlusion no salvageable myocardium is left 81 ; the exact boundaries of such time window are less clear and probably depend on the amount of collateral blood flow, but after more than 12 h coronary occlusion there is little salvageable myocardium left. Patient selection is also mandatory in that patients who really need adjunct protection beyond that by reperfusion per se are recruited into a trial. It is probably not possible to further reduce the less than 3% 1-year mortality which was seen in the neutral ERIC-PPCI/CONDI 2 trial by any cardioprotective intervention.82,83 Then, there are two issues which pertain to better translation on both the preclinical and the clinical side. There is a novel emerging paradigm that not only cardiomyocytes but also the coronary microcirculation are targets of myocardial ischaemia/reperfusion injury18,84,85 and both cardiomyocyte 86 and coronary microvascular injury 87 are determinants of patients’ clinical outcome. The causal and quantitative relationship between infarct size and the area of no-reflow is still unclear, but it is certainly warranted to extend the goal of achieving protection from cardiomyocytes to the coronary circulation. Coronary microvascular obstruction may in fact be a new frontier of cardioprotection. 88 However, a successful attempt to attenuate myocardial ischaemia/reperfusion injury by primarily targeting coronary microvascular obstruction with angiopoietin-like 4 in mice 89 has not been followed up by more translational approaches so far. Another issue that deserves more attention on both the preclinical and the clinical side is the quality and robustness of the methods to assess infarct size. On the preclinical side, the unequivocal gold standard to quantify infarct size is a combination of Evans blue (or a related dye) delineation of area at risk and a triphenyl tetrazolium chloride (TTC) delineation of infarct size; the area of no-reflow is best delineated with thioflavin staining. 90 The gold standard for the assessment of infarct size and area of no-reflow in patients is MRI. 91
Perspective – The Need for Pragmatism and Innovation
So, what would be pragmatic approach to improve the translation of cardioprotection? Notwithstanding the fundamental mechanistic knowledge that can be gained from reductionist approaches, before jumping to clinical application, a given cardioprotective intervention should be tested in a large animal model, such as the pig, with appropriate methodology (TTC) and in a multi-center collaborative approach to assure the robustness of the intervention. The analysis of infarct size data should be performed in a core lab. As a participant in collaborative studies, reviewer or editor I am often appalled by the poor quality of TTC data even from established laboratories when it comes to display of all original data and not only the single best example. The display of all original data in a supplement or separate repository may help to diminish this problem in the future. The CAESAR consortium has pioneered such approach with positive data for ischaemic preconditioning, but nothing else,92,93 and it has unfortunately been discontinued. The IMPACT consortium has made detailed recommendations
94
and is currently repeating the CAESAR approach. It appears also prudent to extend preclinical cardioprotection studies to more long-term outcome as a correlatie to clinical outcome studies such that they cover not only the time of immediate ischaemia/reperfusion injury, but also healing and remodeling.
95
Also, use of human plasma, platelets and myocardial tissue for in vitro studies of cardioprotective effects and signalling is useful before embarking on clinical studies in patients.96–100 Exosomes are released during remote ischaemic conditioning,101–103 may be carriers of cardioprotective signals102,104 and can possibly be exploited as therapeutic vehicles in the future.
105
Very recently, pretreatment with a sodium–glucose transport inhibitor reduced infarct size in isolated perfused mouse hearts,
106
and this approach seems worth to be followed up with more translational approaches. Additive cardioprotection by combining pharmacological
Clearly, cardioprotection has so far not fulfilled its promises to improve patient outcome in clinical practice. This is not so unusual: the field of cell therapy for cardiac repair, which was opened in 2001 by Anversa and colleagues, 112 has attracted many more people and resources than cardioprotection over the years and generated a vast amount of fundamental and novel knowledge in cell biology, but has not even generated a single phase III trial with clinical outcome as endpoint.113,114
We will have to wait and see in the future whether or not a more pragmatic approach to cardioprotection will improve its translation. There is still hope – with more rigor in preclinical studies, better patient selection, focus on the decisive targets (cardiomyocytes and coronary circulation) we may eventually achieve translation of cardioprotection to patient benefit. Of course, innovative science is often disruptive, and the recognition and identification of novel mechanisms and targets of cardioprotection may also bring upon a breakthrough. So, in response to the title's ironic question, we need both: new paradigms and new pragmatism.
Footnotes
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: the German Research Foundation (CRC 1116, B8) and the European Union COST Action of Cardioprotection (CA16225).