
Abstract
G protein-coupled receptors for adenosine (A1, A3, A2A, and A2B), bradykinin (B1) and opioids (δ) are all involved in the mechanism of ischemic preconditioning. Although the heart is comprised of many tissue types, it has been assumed that preconditioning’s protective signaling occurs in the cardiomyocyte. We critically tested that hypothesis by testing for the presence of each of these receptors in isolated adult rabbit ventricular myocytes that had been transfected with cyclic nucleotide-gated (CNG) ion channels. Because subsarcolemmal cyclic adenosine monophosphate (cAMP) opens the CNG channels, we could monitor cAMP levels within a single cardiomyocyte by measuring channel current with a patch pipette. The presence of a receptor would be confirmed if we could alter cAMP in the cell with a selective agonist to the receptor being studied. Superfusion with the β-adrenergic Gs-coupled receptor agonist isoproterenol (50 nmol/L) transiently increased cAMP levels and, therefore, channel current. Pretreatment with selective agonists to A1 or A3 adenosine receptors (ARs) that are Gi-coupled markedly attenuated the response to isoproterenol, indicating inhibition of adenylyl cyclase by increased Gi activity. Agonists to bradykinin or δ-opioid receptors also attenuated isoproterenol’s response. A2AAR and A2BAR are Gs-coupled. The A2AAR–selective agonist CGS21680 increased current through CNG channels but only in the presence of phosphodiesterase (PDE) inhibitors, indicating low surface receptor activity and high intracellular PDE activity. As we previously reported, BAY 60-6583, an A2BAR-selective agonist which mimics preconditioning’s protection in rabbit heart, neither increased nor decreased membrane current in transfected cardiomyocytes, suggesting the absence or a markedly limited number of A2BAR in the sarcolemma. However, reverse transcription polymerase chain reaction (RT-PCR) of purified cardiomyocytes yielded an A2BAR band, implying that rabbit cardiomyocytes do indeed express A2BAR. These data reveal that all receptors reported to be involved in ischemic preconditioning do exist on or within the cardiomyocyte.
Introduction
Preconditioning the heart with a brief period of ischemia (ischemic preconditioning [IPC]) increases the heart’s resistance to infarction from a subsequent prolonged period of ischemia. 1 Ischemic preconditioning is triggered in part by endogenous adenosine which is quickly produced in the ischemic heart and which binds to its Gi protein-coupled receptors (GiPCR), 2 -4 the A1 and A3 adenosine receptor (AR) subtypes. Bradykinin 5,6 and opioids 7 -9 are also released by ischemic myocardium, and they also contribute to IPC’s protection by binding to their respective GiPCR as well. All 4 of these GiPCR act in parallel to trigger entrance into the preconditioned state. Actual protection is thought to occur early in reperfusion when signaling prevents opening of lethal permeability transition pores in mitochondria. 10 We have found evidence that activation of Gs protein-coupled receptor (GsPCR) A2BAR early in reperfusion is required for preconditioning’s protection 11 -13 and a highly selective A2BAR agonist, BAY 60-6583, given at reperfusion mimics IPC’s anti-infarct effect. 13 Finally, activation of the GsPCR A2AAR along with A2BAR has also been proposed to be necessary during reperfusion in the IPC heart. 14
This study critically tests the hypothesis that preconditioning’s signaling pathway occurs in the cardiomyocyte. Although the heart’s ventricle is composed of many cell types (vascular, neural, firoblasts, cardiac muscle, etc), it has been assumed that the abovementioned receptors that are involved in preconditioning reside on the sarcolemma of the ventricular cardiomyocytes, but not all of these receptors have actually been demonstrated in the myocytes. Although many investigators have tried to mimic ischemic or pharmacological preconditioning in isolated cardiomyocytes, the dynamics of ischemic injury and cardioprotection in those models are quite different from those in an intact heart. It is possible that the protective signaling actually occurs in another cell type in the heart and then protects the cardiomyocyte through a diffusible autocoid such as nitric oxide. The cardiomyocyte hypothesis of preconditioning would be disproved if it could be shown that one or more of the receptors used by IPC’s signaling are not expressed by the cardiomyocyte. Adenosine A1AR stimulation markedly inhibits isoproterenol (iso)-induced increase in contractility 15 as does bradykinin, 16 indicating that both of those receptors coexist with adrenergic receptors on cardiomyocytes. It has been more difficult to demonstrate the presence of the remaining receptors. An A2AAR agonist reportedly modestly enhances cardiac contractility. 17 However, that could have been an artifact resulting from A2AAR-mediated dilation of the coronary vasculature. 18 There is even less evidence that A3AR or opioid receptors are present on the cardiomyocyte.
We have recently presented histological evidence that A2BAR are expressed by the rat cardiomyocyte but reside on or near the mitochondria and likely not in the cell’s sarcolemma. 19 We subsequently tested for the functional presence of A2BAR in single rabbit cardiomyocytes with a technique that monitors intracellular cyclic adenosine monophosphate (cAMP) levels. 20 Isolated ventricular myocytes were transfected with cyclic nucleotide-gated (CNG) channels. Increasing the concentration of cAMP near the sarcolemma opens the channel which in turn causes increased membrane current that can be measured with a whole-cell patch clamp technique. Cationic current through CNG channels has been shown to be proportional to cAMP concentration. 21 Activation of a GsPCR stimulates adenylyl cyclase activity and raises cAMP, while activation of a GiPCR inhibits adenylyl cyclase activity and lowers cAMP. These changes in cAMP are then reflected in the magnitude of the membrane current measured with a patch pipette. This method has been shown to have high resolution for subsarcolemmal cAMP. 22,23 Consistent with our immunohistochemistry findings that A2BAR are on or near mitochondria, 19 we failed to alter membrane current with an A2BAR-selective agonist, confirming that rabbit cardiomyocytes do not have A2BAR in their sarcolemma. 20 In the present study, we used this same model to test whether any of the remaining receptors associated with IPC could be found in the rabbit cardiomyocyte.
Materials and Methods
Isolation and Culture of Rabbit Ventricular Myocytes
New Zealand White rabbits of either gender were used. The Institutional Animal Care and Use Committee of the University of South Alabama College of Medicine approved these protocols. All animals were cared for and handled in accordance with the guidelines established by the National Institutes of Health.
24
Rabbit ventricular myocytes were isolated as described previously.
25
Briefly, a rabbit heart was mounted on a Langendorff apparatus and retrogradely perfused with calcium-free Krebs–Henseleit–HEPES buffer containing collagenase. The softened heart was minced and extruded through fine nylon mesh. Myocytes were made calcium tolerant by stepwise restoration of calcium. Cells were plated on laminin-coated glass cover slips in 2 mL Medium-199 modified with Earle’s salts containing
Measurement of cAMP Signals in Single Myocytes
Cyclic AMP signals near the plasma membrane were measured using genetically modified CNG channels as described previously.
21,26,27
Myocytes were transfected using adenovirus-encoding CNGA2 subunits with the C460W and E583M mutations. These channels have an apparent affinity for cAMP of 1.1 μmol/L, allowing the measurement of near-membrane-free cAMP levels of 0.1 to 10 μmol/L.
28
Binding of cAMP to CNG channels triggers a conformational change in the channel, allowing cation flux through the channels thus producing electric currents. Currents were measured with the whole-cell patch clamp technique. Patch pipettes were filled with buffer containing (in mmol/L) 140 KCl, 10 HEPES, 5 Na2ATP, 0.5 Na2GTP, and 0.5 MgCl2 at pH 7.4. The perfusion buffer contained (in mmol/L) 145 NaCl, 4 KCl, 10 HEPES, 10
Determination of the Presence of GPCRs in Myocytes
Protocol 1 (Figure 1) was used for each receptor agonist to determine whether Gi or Gs protein-coupled receptors were present. The cell was bathed for 3 minutes in buffer to establish a baseline. Addition of an agonist of one of the heart’s GsPCRs would be expected to activate adenylyl cyclase and increase membrane current. If there were no response to the agonist alone, then 50 nmol/L iso was infused in addition to the agonist to raise cAMP and test whether the cell was viable and the peak current (i p) was noted. If the agonist instead activated GiPCRs, then it should reduce cAMP and membrane current. Resting rabbit cardiomyocytes have high PDE activity, causing cAMP to be below the threshold for channel opening. Hence, further lowering of cAMP would not be detected. We, therefore, tested whether the agonist could attenuate the response to 50 nmol/L iso. At the end of the experiment, PDEs were inhibited in the presence of iso to establish the maximal iso-induced current (i max). If the agonist significantly decreased i p/i max compared to that seen with iso alone, then we concluded the agonist had activated GiPCR.

Experimental protocols. The horizontal axis is time. Protocol 1 is used to test for the functional presence of a Gs protein-coupled receptor with a highly selective agonist for that receptor. The subsequent addition of isoproterenol as a positive control tests whether the cell is viable and expressing the CNG channels. Finally, the PDE inhibitors reveal the level of PDE activity in the cell. If activity is not seen in protocol 1, then protocol 2 is used. By starting the PDE inhibitor prior to the agonist, we can test whether high PDE activity had masked the increase in cAMP expected following occupancy of the receptor in protocol 1. EC, extracellular solution; PDE, phosphodiesterase; cAMP, cyclic adenosine monophosphate.
The cardiomyocyte’s high-basal PDE activity could keep cAMP from rising in response to a GsPCR agonist if the receptor density was very low. 29 Therefore, if protocol 1 did not reveal receptors to be either Gs or Gi coupled, we tested for PDE masking with protocol 2 (Figure 1). The PDE inhibitor cocktail causes cAMP to rise, revealing a high-background adenylyl cyclase activity. After several minutes, the cAMP level begins to subside, presumably because of feedback inhibition of adenylyl cyclase activity as well as residual PDE activity. 26,27,30 The GsPCR agonist is then added during this declining phase. With the inhibition of PDE activity, even a small increase in adenylyl cyclase activity should be observable. Concentrations of agonists were approximately 10-fold their published K ds.
Reverse Transcription–Polymerase Chain Reaction (RT-PCR) of Cardiomyocytes for Adenosine A2B Receptor Message
RNA was extracted from cardiomyocytes using the RNeasy fibrous tissue kit from QIAGEN. In brief, the myocytes were digested with Proteinase K, and the supernatant was passed through the RNeasy Mini spin column. After RNase-free DNase digestion, the RNA pellets were washed off the column in nuclease-free water by centrifugation and stored at −80°C. RNA was reverse transcribed using the iScript complementary DNA (cDNA) Synthesis kit from Bio-Rad. The resulting cDNA was amplified by PCR with primers designed to span one intron region and electrophoresed through 3% agarose gels. The primers for amplification of the 300-bp rabbit gylceraldehyde 3-phosphate dehydrogenase (GAPDH) were as follows: forward primer = GAT GGT GAA GGT CGG AGT G and reverse primer = AAG ACG CCA GTG GAT TCC AC. The primers for amplification of the 273-bp rabbit adenosine A2BAR were forward primer = CTG CTT CGT GCT TGT GCT and reverse primer = AAC AGA CAC TTG ACG AGG CA. The primers for amplification of the 235-bp A1AR were forward primer = GCC ACC TTC TGC TTC ATC G and reverse primer = GCG TCA CCA CTG CCT TGT A. All primers were designed using Primer 5.0 software.
Three groups of cardiomyocytes were studied. First, we used the crude cardiomyocyte preparation described above, which is obviously contaminated with other cell types. The second group was myocytes labeled with tetramethylrhodamine methyl and ethyl esters (TMRE). To purify the myocytes, cells with high fluorescence were sorted with a Becton-Dickinson FACSVantage SE flow cytometer. The third group consisted of several hundred myocytes which were individually picked up with a manually guided micropipette under a microscope.
Statistics
All values are reported as mean ± standard error of the mean (SEM). Differences between groups were determined by 1-way analysis of variance and Student-Newman-Keuls post hoc test. P < .05 was considered significant.
Results
Iso-Triggered cAMP Signals in Single Myocytes
In rabbit cardiomyocytes transfected with CNG channels, 50 and 100 nmol/L iso caused dose-dependent transient increases of cAMP monitored as whole-cell current (Figure 2A and B). Adding a PDE inhibitor cocktail increased current to a plateau which we considered to be maximal current (i max). The ratio of iso-triggered peak current (i p) to maximal current (i max), that is, i p/i max, averaged 0.56 ± 0.09 (n = 5) for 50 nmol/L iso and 0.72 ± 0.10 (n = 5) for 100 nmol/L iso (Figure 2C). Because 50 nmol/L iso caused a submaximal response, we chose that dose for the remaining protocols. Using our protocol, iso caused a negligible change in current in cardiomyocytes not transfected with CNG channels (n = 8; data not shown).

Typical isoproterenol (iso)-triggered cAMP signals in rabbit ventricular myocytes measured as currents through cyclic-nucleotide gated (CNG) channels. (A) 50 and (B) 100 nmol/L iso-triggered transient increases in cAMP levels. Subsequent addition of a phosphodiesterase (PDE) inhibitor cocktail increased currents to a plateau, which is the iso-induced maximal current (i max). Average ratio of iso-triggered peak current (i p) to i max in the presence of the PDE inhibitor cocktail for both groups is presented in panel (C). Numbers in parentheses indicate the number of cells studied and the bars represent SEM. cAMP indicates cyclic adenosine monophosphate; SEM, standard error of the mean.
GiPCR Agonists Attenuate Iso-Triggered cAMP Signals
A GsPCR agonist such as iso stimulates adenylyl cyclase activity which increases synthesis of cAMP. Simultaneous activation of GiPCRs would compete with that activation and reduce cAMP production. We determined the expression of GiPCRs like A1AR on cardiomyocytes by documenting their ability to attenuate the response to isoproterenol. Myocytes were pretreated with the highly selective A1AR agonist 2-chloro-N 6-cyclopentyladenosine (CCPA, 200 nmol/L) 31 using protocol 1 (Figure 1). 2-Chloro-N 6-cyclopentyladenosine had no effect by itself but clearly attenuated the iso-induced rise in cAMP (Figure 3A). Washout of CCPA was always accompanied by a transient increase in cAMP as Gi-mediated inhibition was removed (Figure 3A). 2-Chloro-N 6-cyclopentyladenosine significantly decreased the average i p/i max for iso (P < .025; Figure 4).
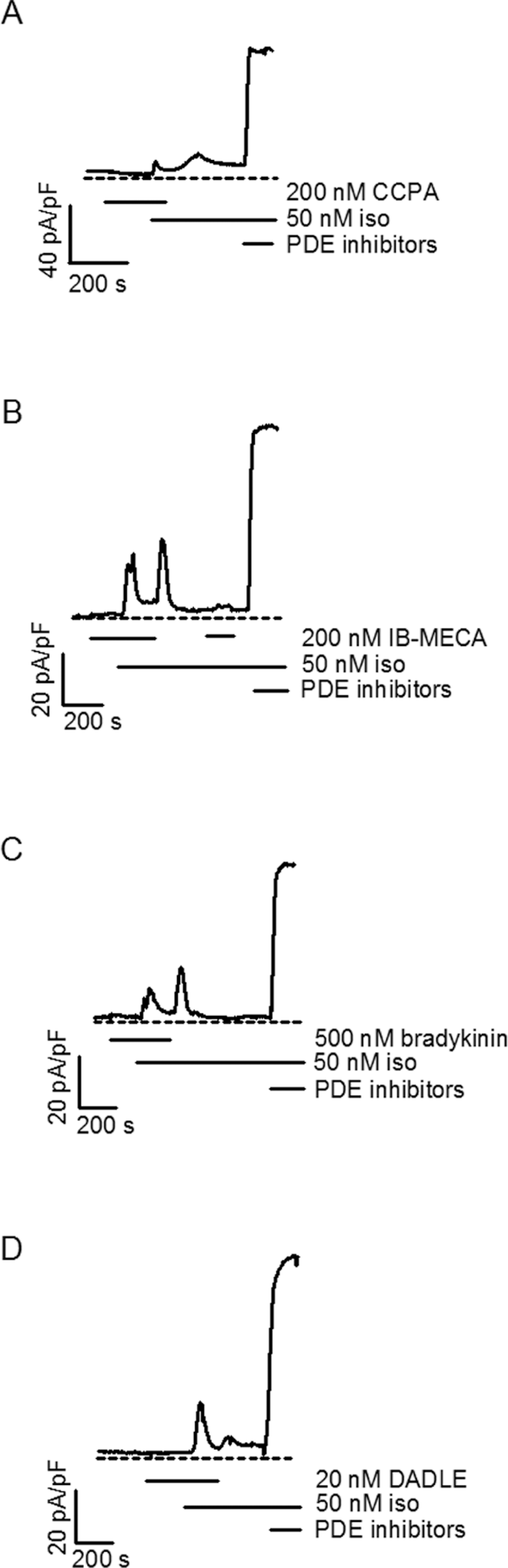
Activation of adenosine A1 (A), adenosine A3 (B), bradykinin (C), and δ-opioid (D) receptors inhibit isoproterenol (iso)-triggered cAMP signals in rabbit cardiomyocytes. In each case, washout of the agonist revealed a second increase in cAMP as the inhibitory effect of Gi activity on adenylyl cyclase was removed. CCPA, 2-chloro-N
6-cyclopentyladenosine; DADLE, (

Ratio of peak isoproterenol (iso) current to the maximum current observed after inhibition of phosphodiesterases (i
p/i
max) is plotted for 50 nmol/L iso alone and iso plus each of the 5 agonists tested with protocol 1. Numbers of cells tested are indicated in parentheses. The first 4 agonists caused a significant decrease in the iso response but BAY 60 did not. BAY 60, BAY 60-6583; CCPA, 2-chloro-N
6-cyclopentyladenosine; DADLE, (
A3AR is another GiPCR associated with IPC. 4 The A3AR-selective agonist N 6-(3-iodobenzyl)-adenosine-5′-N-methylcarboxamide (IB-MECA, 200 nmol/L) 32 also significantly attenuated the iso-triggered cAMP signal and washout of IB-MECA caused cAMP to rise again (Figure 3B). The average i p/i max for iso was significantly decreased as well (P < .02; Figure 4).
Bradykinin B2 and δ-opioid receptors are Gi-coupled and both, like A1AR and A3AR, trigger a preconditioned state.
5,6,9,33
We sought to determine whether their receptors could be detected on rabbit cardiomyocytes. Pretreatment with 500 nmol/L bradykinin either completely abolished or greatly attenuated iso’s effect on cAMP (Figure 3C). The average decrease was highly significant (P < .005; Figure 4). The δ-opioid-selective agonist ([
A2AAR and A2BAR—GsPCRs
It is controversial whether functional A2AR reside in or on cardiomyocytes. A2AAR reportedly couple to Gs. 34 The highly A2A-selective agonist CGS 21680 (300 nmol/L) alone did not change the basal cAMP level and no current was detected. Addition of PDE inhibitor cocktail in the presence of CGS 21680 caused a large increase in measured current, indicating that adenylyl cyclase activity had been masked by the PDEs (Figure 5A). However, this protocol could not determine whether A2AAR had contributed to that activity. According to protocol 2 (Figure 1), the cell was first treated with the PDE inhibitor cocktail which increased the current and revealed a high adenylyl cyclase activity even without receptor stimulation. That increase in cAMP was transient, however, and the current slowly declined, presumably because of feedback inhibition of adenylyl cyclase and increased PDE activity. 26,27,30 Adding CGS 21680 during that decline caused a clear increase in current (Figure 5B). Therefore, a small but detectable population of functional A2AAR on cardiomyocytes could be uncovered but only when PDE activity was inhibited.

Adenosine A2 receptor-mediated cAMP signals in rabbit cardiomyocytes. A, The A2A-selective agonist CGS 21680 did not change the basal cAMP level before application of phosphodiesterase (PDE) inhibitors (n = 3). B, PDE inhibitor cocktail transiently increased cAMP and addition of the A2A agonist while cAMP level was declining, presumably because of feedback inhibition of adenylyl cyclase, could then increase adenylyl cyclase activity in face of the effective PDE blockade and elevate cAMP (n = 5). Thus, a small but measurable expression of A2AAR was found. However, the A2BAR-selective agonist BAY 60-6583 (Bay 60) did not have any effect on cAMP whether introduced before (C) (n = 5) or after (D) (n = 4) the PDE inhibitor cocktail. iso indicates isoproterenol; cAMP, cyclic adenosine monophosphate.
A2BAR are also reported to be Gs coupled. 34 We recently reported that BAY 60-6583, a highly A2B-selective agonist, did not change membrane current in the transfected myocytes even in the presence of PDE inhibitors. 20 Representative data from those experiments are shown in Figure 5C and 5D. To test whether these receptors might possibly be Gi coupled, we investigated whether pretreatment with 1 μmol/L BAY 60-6583 would attenuate iso-triggered cAMP signals. It did not (Figure 4). Thus, there was no sign of either Gs or Gi action of BAY 60-6583, indicating that the A2BAR density is too low to influence subsarcolemmal cAMP levels in rabbit cardiomyocytes. When BAY 60-6583 was given to HEK 293 cells transfected with human A2BAR and the CNG channels, it caused increased membrane current in a dose-dependent manner. 20
Reverse Transcription–Polymerase Chain Reaction
Single-cell PCR revealed that rat cardiomyocytes express A2BAR, 19 but it is unknown whether rabbit cardiomyocytes express them. Figure 6 shows the results of the RT-PCR studies. The crude myocyte preparation showed bands for both A1AR and A2BAR. However, we know that this preparation is contaminated with other cell types including fibroblasts and coronary vascular cells, and hence we could not unequivocally attribute the receptor bands to the cardiac myocytes. We tried to purify the cardiomyocytes by sorting them for TMRE fluorescence with our flow cytometer. The culture was labeled with TMRE which concentrates in healthy mitochondria which are polarized. Because cardiomyocytes are both large and rich in mitochondria, they were easy to identify by their bright fluorescence. Although the A2BAR message was reduced, it was still detectable. We still were not satisfied that our culture was 100% cardiomyocytes so we hand selected approximately 200 myocytes from the fluorescence-activated cell sorting (FACS)-sorted cells using a micropipette and a microscope. The A2BAR band could still be seen in the hand sorted cells. Therefore, our results indicate that cardiomyocytes do express A2BAR, but, like the case in rat cardiomyocytes, 19 they do not appear to be in the sarcolemma.
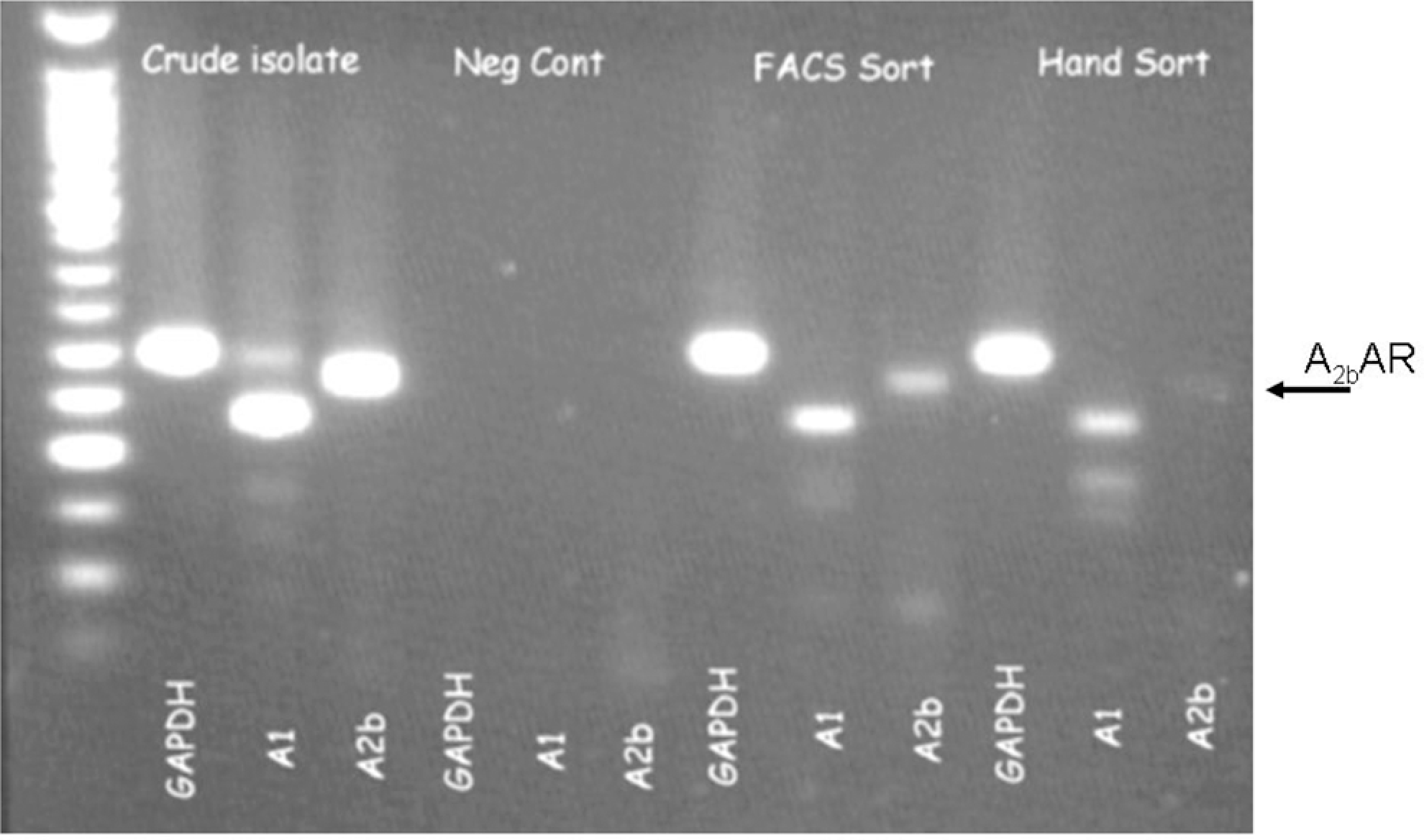
Reverse transcription–polymerase chain reaction (RT-PCR) of message for adenosine gylceraldehyde 3-phosphate dehydrogenase (GAPDH), and A1 and A2B receptors from crude and purified rabbit cardiomyocyte preparations. See text for details.
Discussion
We found clear evidence for the functional expression of 5 of the 6 receptors associated with IPC’s protective pathway in the cardiomyocyte’s sarcolemma. We have shown that A3AR are involved in preconditioning in the rabbit heart. 4 The A3-selective agonist Cl-IB-MECA was able to induce protection against hypoxia in a neonatal rat cardiomyocyte model, 35 but there are little available data confirming whether A3AR are actually expressed in adult cardiac myocytes. Zhang et al 36 found that A1AR-selective agonists inhibited high dihydroouabain-sensitive Na+-K+ current in guinea pig ventricular myocytes, whereas Cl-IB-MECA had no effect causing them to conclude that adult cardiomyocytes do not express A3AR, at least in that species. The present results, however, clearly reveal A3AR in the sarcolemma of adult rabbit cardiomyocytes. It is well known that cardiomyocytes express A1AR since the activation of A1AR has a marked antiadrenergic effect on the rat heart, 37 which is consistent with our observation that A1AR are in the sarcolemma. Bradykinin also competes with adrenergic stimulation of the heart. 16
Our rabbit cardiomyocytes have surprisingly high basal adenylyl cyclase activities, resulting in a futile cycling of cAMP synthesis and breakdown in these quiescent cells. This basal adenylyl cyclase activity is reflected in the rise in cAMP when the major PDEs were inhibited with the PDE inhibitor cocktail. As a result, we had to inhibit PDEs before we could demonstrate the presence of A2AAR. We cannot determine whether the high-PDE activity is representative of cells in the intact heart or if it is induced by our cell culture conditions because it takes 48 hours in culture to express CNG channels. Our results are consistent with the previous observations in which inhibition of PDE activity alone increased cAMP levels in neonatal rat cardiac myocytes. 38 Phosphodiesterase inhibition alone also potentiated protein kinase A (PKA)-mediated L-type Ca2+ channel activity 39 and total cAMP content 40 in adult rat cardiomyocytes. But unlike the present study, inhibition of PDE activity alone induced little or no increase in CNG channel activity in adult rat cardiomyocytes, possibly due to buffering of free cAMP by PKA or PKA-mediated effects on PDE and adenylyl cyclase activities. 41 Yet PDE inhibition still greatly potentiated iso-induced cAMP signals. These observations suggest that basal PDE and adenylyl cyclase activities may vary substantially between species.
A2AAR-selective agonists have a modest positive inotropic effect in rat 42 and chick 43 hearts, suggesting their presence on cardiomyocytes but that could have been an artifact from coronary dilation by the agonist. Our findings confirm A2AAR activity in isolated rabbit heart muscle cells. Unlike the case with iso, PDE inhibition was required to observe an A2AAR agonist-induced increase in cAMP in cardiac myocytes. Yet the concentration of CGS21680 that we used should have occupied nearly 100% of A2AARs. The difference was likely due to different densities of β-adrenergic and A2AARs on the sarcolemma but could also reflect some compartmentalization of cAMP.
We 13,44 and others 45 have found the A2BAR to be an important part of IPC’s protective pathway. It has been established that A2BARs are present in coronary vessels where they cause dilation. 46 They are also present in cardiac fibroblasts. 47,48 As a result, cardiac biopsies yield message for A2BAR but that does not prove that they are expressed in cardiomyocytes. Reverse transcription–polymerase chain reaction revealed message for A2BAR in isolated rat cardiomyocytes 19 ; and in the present study, we were able to show message for A2BAR in highly purified rabbit cardiomyocytes. We were unable to show any effect on cAMP when the myocytes were treated with BAY 60-6583, an A2BAR-selective agonist, and that was assumed to be related to their apparent intracellular location. 19 Receptors on or near the mitochondria would likely not affect the subsarcolemmal cAMP pool that controls the CNG channels. The intracellular receptors do appear to be functional in that we could inhibit rotenone-induced superoxide production in isolated rabbit cardiomyocytes with BAY 60-6583 and that could be blocked by an A2BAR-selective antagonist MRS 1754. 20
In summary, we were able to demonstrate receptor-mediated changes in cAMP for A1AR, A2AAR, and A3AR as well as bradykinin and δ-opioid receptors in single rabbit ventricular cardiomyocytes and genetic evidence of A2BAR in isolated ventricular myocytes, thus supporting the hypothesis that preconditioning’s signaling occurs wholly within the heart’s cardiomyocytes.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The project described was supported by award numbers HL-20468 and HL-74278 from the National Heart, Lung, and Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health. BAY 60-6583 was a generous gift from Thomas Krahn of Bayer AG.