
Abstract
Myocardial ischemia has become one of the main causes of sudden cardiac death worldwide. Autophagy has been demonstrated to protect cardiomyocytes from ischemia/reperfusion (I/R)-induced damage. A novel small molecule compound 2-Chloro-5-[[5-[[5-(4,5-Dimethyl-2-nitrophenyl)-2-furanyl]methylene]-4,5-dihydro-4-oxo-2-thiazolyl]amino]benzoic acid (PT1) has been previously shown to specifically activate 5′-adenosine monophosphate-activated protein kinase (AMPK). Because AMPK activation effectively induces autophagy, we tested the protective efficacy of PT1 on cardiomyocytes after oxygen glucose deprivation/reoxygenation (OGD/R) in vitro. Mouse neonatal cardiomyocytes were treated with PT1 after OGD/R. 3-[4-(1,3-benzodioxol-5-yl)-2-oxo-3-buten-1-yl]-3-hydroxy-1,3-dihydro-2H-indol-2-one (3HOI-BA-01), a novel small compound showing potent inhibitory effect on mammalian target of rapamycin (mTOR) activation, was also tested for its cardioprotective effect, based on the established relationship between mTOR signaling and autophagy. Cell survival and autophagy-related signal pathways were examined after treatment with these agents. Our data indicate that both PT1 and 3HOI-BA-01 enhance cell survival after OGD/R. As expected, both PT1 and 3HOI-BA-01 induced autophagy in cardiomyocytes through activating AMPK pathway and inhibiting mTOR signaling, respectively. Induction of autophagy by PT1 and 3HOI-BA-01 was responsible for their cardioprotective effect, since inhibition of autophagy abolished the protective efficacy. Furthermore, simultaneous administration of PT1 and 3HOI-BA-01 profoundly upregulated autophagy after OGD/R and significantly promoted survival of cardiomyocytes. In vivo administration of PT1 and 3HOI-BA-01 in a murine myocardial (I/R injury model remarkably reduced infarct size and induced autophagy. Taken together, our research suggests that PT1 and 3HOI-BA-01 could be promising therapeutic agents for myocardial ischemia.
Introduction
Myocardial ischemia has become one of the leading causes of sudden cardiac death over the last few decades. 1 Alleviation of ischemia/reperfusion (I/R)-induced myocardial injury has therefore attracted much attention. This is primarily due to the limited ability of the injured heart tissue to regenerate through proliferation of pre-existing cardiomyocytes. 2 The mechanisms underlying ischemic damage in cardiomyocytes have not been fully elucidated. Molecular and biochemical events following ischemic stress are complicated and include the interaction of multiple biological signal pathways. 3 Increased intracellular calcium and sodium, dissipation of mitochondrial membrane potential, free radical formation/reactive oxygen species, dysregulated nitric oxide (NO) metabolism, endothelial dysfunction, platelet aggregation and microembolization, immune activation, and apoptosis are all involved in postischemic injury. 4 –6
Autophagy has been a focus of research since it was discovered decades ago. Emerging evidence suggests that autophagy is a stringently regulated process, which plays critical roles in cell growth, differentiation, and homeostasis, as well as in maintaining a balance between the synthesis, degradation, and recycling of cellular products. 7 –9 Recent studies indicate that autophagy is also crucial for maintenance of homeostasis in the heart. 9,10 Furthermore, autophagy plays an important role in the regulation of cardiomyocyte survival after ishemia/reperfusion. 11,12 Among several signal transduction pathways, which are crucial for induction and regulation of autophagy, the mammalian target of rapamycin complex 1 (mTORC1) pathway and the 5′-adenosine monophosphate-activated protein kinase (AMPK) pathway are considered the key modulating pathways. mTOR, regulatory-associated protein of mTOR (Raptor), mammalian lethal with SEC13 protein 8 (MLST8), and Proline-rich-Akt Substate of 40kDa (PRAS40) and DEP domain containing MTOR-interacting protein (DEPTOR) form mTORC1. mTORC1 phosphorylates eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) and p70S6 kinase to initiate protein synthesis. mTORC1 signaling acts as a negative regulator for autophagy by inhibiting Ulk1, which is critical for membrane nucleation. AMPK signaling acts as a master regulator of cellular energy homeostasis. AMPK is activated in response to stresses that deplete cellular adenosine-5′-triphosphate (ATP) supplies such as low glucose, hypoxia, ischemia, and heat shock. AMPK signaling regulates autophagy directly through phosphorylation of Ulk1 or indirectly through inhibiting mTOR signaling. 8,13
2-Chloro-5-[[5-[[5-(4,5-Dimethyl-2-nitrophenyl)-2-furanyl]methylene]-4,5-dihydro-4-oxo-2-thiazolyl]amino]benzoic acid (PT1) is a novel AMPK activator. It dose-dependently activates AMPK α1394, α1335, α2398, and even heterotrimer α1β1γ1. Based on the PT1-docked AMPK α1 subunit structure model and different mutations, it is speculated that PT1 interacts with Glu-96 and Lys-156 residues near the autoinhibitory domain and directly relieves autoinhibition. 14,15 Studies using L6 myotubes showed that the phosphorylation of AMPK and its downstream substrate, acetyl-coenzyme A carboxylase (ACC), are dose-dependently and time-dependently increased by PT1 without an increase in cellular 5′-adenosine monophosphate (AMP):ATP ratio. 14 In Hela cells, PT1 enhances AMPK phosphorylation, which can be inhibited by the AMPK inhibitor compound C. 14 However, the effect of PT1 on AMPK signaling in cardiomyocytes has not been studied. Moreover, whether PT1-induced AMPK activation effectively promotes autophagy is unknown. Previous studies confirmed that AMPK signaling modulates cell autophagy and metabolism under nutrient deprivation conditions. 16,17 Therefore, we speculated that PT1 could be used for treating cardiomyocytes after I/R, in which autophagy has been proven to be protective. 3-[4-(1,3-benzodioxol-5-yl)-2-oxo-3-buten-1-yl]-3-hydroxy-1,3-dihydro-2H-indol-2-one (3HOI-BA-01) is a novel mTOR inhibitor. Its antitumor efficacy has recently been studied both in vitro and in vivo. 18 The in vitro study demonstrated that 3HOI-BA-01 inhibited mTOR kinase activity by directly binding to mTOR. However, its efficacy for myocardial ischemia has not been reported. Other mTOR inhibitors, such as rapamycin, have been demonstrated to be cardioprotective in several cases. 19 –21 Therefore, research on the effect of 3HOI-BA-01 in protecting cardiomyocytes after oxygen glucose deprivation/reoxygenation (OGD/R) would provide clues for novel therapeutic approaches for preventing and treating cardiac ischemia.
In this study, we used in vitro and in vivo models to test whether PT1 and 3HOI-BA-01 could protect mouse cardiomyocytes after ischemia. Our results indicate that both agents are capable of enhancing cell survival after OGD/R in vitro. Simultaneous administration of both compounds induced the most protective effect. Furthermore, we demonstrated that their protective effect is attributed to induction of autophagy through activation of the AMPK pathway and inhibition of mTOR signaling, respectively. Injection of PT1 with 3HOI-BA-01 in a mouse myocardial ischemia model produced cardioprotection after ischemic damage in vivo. Taken together, our research could provide a novel approach to treat myocardial ischemia.
Materials and Methods
Mouse Primary Cardiomyocyte Culture and OGD/R
The protocol was approved by the Wuhan University School of Medicine Animal Care and Use Committee. All animals received treatment in compliance with institutional guidelines and Wuhan University Guidelines for the Use of Animals. C57BL/6 mice were purchased from Beijing HFK Bioscience Co Ltd (Beijing, China).
Primary cardiomyocyte cultures were prepared from ventricles of 1-day-old C57BL/6 mice according to published methods with a few modifications. 22,23 Briefly, neonatal mice were anesthetized with pentobarbital sodium (50 mg/kg intraperitoneally). The hearts were removed aseptically. The ventricles from 3 to 6 hearts were pooled. Ventricles were kept on ice in Hanks’ balanced salt solution (HBSS) without Ca2+ and Mg2+. The ventricles were then washed 3 times with HBSS and minced into small fragments. Cells were dissociated at 25°C for 15 minutes with 0.625% trypsin in HBSS without Ca2+ and Mg2+. Cells released after the first digestion were discarded. Cells from subsequent digestions were added to an equal volume of cold HBSS with Ca2+ and Mg2+ until all cardiac cells were isolated. The mixture was centrifuged at 200g for 10 minutes, and the cells were resuspended in Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F-12 nutrient mixture (Sigma-Aldrich, St Louis, Missouri) supplemented with estrogen-free 5% fetal bovine serum (Hyclone, Logan, Utah), 100 U/mL penicillin, 100 μg/mL streptomycin, and 25 μmol/L cytosine arabinoside. To exclude nonmuscle cells, the isolated cells were first plated in tissue culture dishes at 37°C for 1 hour in an incubator with 5% CO2–95% O2. Suspended cardiomyocytes were then collected and plated at a density of 1.0 × 105 cells/cm2 and incubated under the same conditions as above for 24 hours.
For OGD stimulation, OGD medium, that is, HBSS with Ca2+ and Mg2+ free of serum and glucose was pre-equilibrated in 100% N2 at 37°C for 2 hours. Oxygenated medium was removed from the cardiomyocyte cultures, and the N2-pre-equilibrated glucose-free medium was immediately added. Cultures were promptly placed in a hypoxia chamber and exposed to 100% N2 for 1.5 hours at 37°C. For reoxygenation, the glucose-free medium was replaced with supplemented DMEM/Ham’s F-12, and the cells were reoxygenated in 21% O2–5% CO2–74% room air for 3 hours at 37°C.
PT1 and 3HOI-BA-01 Treatment
PT1 was purchased from Tocris Bioscience (Brisol, United Kingdom). PT1 stock solution was prepared by dissolving PT1 in dimethyl sulfoxide (DMSO) at 100 mmol/L and was stored at −20°C before use. 3HOI-BA-01 was purchased from ChemBridge (San Diego, California). Its stock solution was prepared by dissolving 3HOI-BA-01 in DMSO at 10 mmol/L and was stored at −20°C. For testing their cytotoxic or effective dosage, the stock solution was diluted with cardiomyocyte culture medium to indicated concentrations immediately before use. Twenty four hours after seeding cells, the culture medium was discarded and cells were gently rinsed with phosphate-buffered saline (PBS). The agent-containing culture medium was then added into each well and cells were incubated for additional 24 hours.
For treating cells after OGD/R, PT1 and 3HOI-BA-01 were diluted in supplemented DMEM/Ham’s F-12 before use. OGD/R-treated cells were incubated in PT1- and/or 3HOI-BA-01-containing medium in 21% O2–5% CO2–74% room air for another 24 hours at 37°C. In some experiments, the AMPK inhibitor compound C (Sigma-Aldrich) or phosphatidylinositol-3-kinases (PI-3K) inhibitor pervanadate (Sigma-Aldrich) was used at indicated concentrations in the presence of PT1 or 3HOI-BA-01 to treat cells.
For in vivo induction of autophagy, 100 mg/kg PT1 and 100 mg/kg 3HOI-BA-01 were administered through intraperitoneal injection into 8 to 12-week-old male C57BL/6 mice once a day for 3 consecutive days. Then mice were sacrificed by CO2 inhalation and hearts were collected and homogenized in radioimmunoprecipitation assay (RIPA) buffer with homogenizers. Tissue was lysed in RIPA buffer on ice for 30 minutes and lysates were stored at −80°C before analysis.
Cell Viability Assay
Cell viability assay was performed using Calcein-AM (R&D Systems, Minneapolis, Minnesota) following the manufacture’s instruction. Briefly, cells were grown in 96-well plates before the assay. Culture medium was carefully discarded and cells were gently washed with PBS twice. 1 µmol/L Calcein-AM was prepared by diluting Calcein-AM in prewarmed PBS and was added into each well. Cells were incubated for 30 minutes in a 37°C incubator before recording fluorescence on a Tecan Infinite 200 PRO plate reader (Tecan Systems, San Jose, California).
Flow Cytometry
Floating cells were collected after treatment. Adherent cells were incubated in 2 mmol/L EDTA–PBS at 37°C for 10 minutes. Cells were gently flushed with a pipette to dissociate cells from the bottom of wells. Flushed cells were pooled with floating cells. Cells were then centrifuged and stained with propidium iodide (BD Biosciences, San Jose, California) and Annexin V (BD Biosciences) according to the manufacture’s instruction. Briefly, cells were washed with PBS twice and incubated in 100 µL of binding buffer (10 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.4; 140 mmol/L sodium chloride (NaCl); and 2.5 mmol/L calcium chloride) containing Annexin V–fluorescein isothiocyanate (1:20) and 5 µg/mL propidium iodide for 15 minutes at room temperature (RT) in the dark. Four hundred microliters of binding buffer was added to each sample. Apoptosis was analyzed by flow cytometry within 1 hour.
Western Blot
Cell lysate was prepared by homogenization in RIPA buffer (20 mmol/L Tris–hydrochloric acid, pH 7.5, 150 mmol/L NaCl, 1 mmol/L disodium EDTA, 1 mmol/L ethylene glycol tetraacetic acid, 1% nonyl phenoxypolyethoxylethanol, 1% sodium deoxycholate) and lysed on ice for 30 minutes. The amount of protein was quantified using Pierce 660 nm Protein Assay (Thermo Scientific, Waltham, Massachusetts) following the manufacture’s instruction. Total protein from each sample was loaded onto 8% (for ACC) or 10% (for extracellular signal-regulated kinase (Erk), AMPKs, B-cell lymphoma 2 (Bcl-2), and β-actin) sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel and electrophoresis was performed. For light chain 3 isoform B (LC3B), proteins were loaded onto 4%–20% Mini-PROTEAN® TGX™ Precast Gels (Bio-Rad, Hercules, California). Proteins were then transferred onto nitrocellulose membranes and blocked with 5% nonfat dry milk–PBS for 1 hour at RT. After 3 washes with 0.05% Tween-20–PBS (PBST), membranes were incubated with the following primary antibody reagents overnight at 4°C: (1) rabbit monoclonal anti-AMPKα, anti-phospho-AMPKα, anti-ACC and anti-phospho-ACC, anti-Erk1/2, and anti-phospho-Erk1/2 (Cell Signaling Technology, Beverly, Massachusetts) and (2) mouse monoclonal anti-Bcl-2 and anti-β-actin (Santa Cruz Biotechnology, Santa Cruz, California). Membranes were washed 3 times with PBST and then incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hour at RT. Membranes were developed with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and were visualized and the optical density of the identified protein bands on membranes was analyzed using a Bio-Rad ChemiDoc MP system.
Immunocytochemical Staining
Cells were seeded in each well of 8-well Lab-Tek™ chamber slides (Thermo Scientific) and were treated as described earlier. Cells were then fixed with 4% paraformaldehyde and stained with rabbit anti-LC3B antibody (Cell Signaling Technology) and rat anti-lysosomal-associated membrane protein-1 (LAMP-1) antibody (Biolegend, San Diego, California) followed by incubation with Alexa Fluor 594-conjugated goat anti rabbit immunoglobulin G (IgG) and Alexa Fluor 488-conjugated goat anti rat IgG (both from Invitrogen, Carlsbad, California). Fluorescent microscopy was conducted using a Leica DM4000 B light-emitting diode microscope (Leica Microsystems, Wetzlar, Germany).
Mouse Myocardial I/R Model
Male C57BL/6 J mice at 8 to 10 weeks of age were used for the surgical procedure, which was performed according to the Guiding Principles in the Care and Use of Animals of the American Physiological Society and was in accordance with the Guide for the Care and Use of Laboratory Animals. Mice were anesthetized with 2% halothane (Sigma-Aldrich) and 40% oxygen, and maintained with 0.5% halothane and 40% oxygen. Tracheotomy was performed to provide artificial ventilation (0.3 mL tidal volume, 120 breaths/min), and the left coronary artery was ligated with an 8-0 nylon surgical suture 1.0 mm distal from tip of the left auricle. Sham-operated animals underwent the same surgical procedure except that the suture was not tied around the left anterior descending (LAD) artery. Following 1-hour LAD occlusion, PT1 and 3HOI-BA-01 were intraperitoneally injected once a day for 3 consecutive days. After 1-hour ligation and 3-day reperfusion, the ventricles were frozen and cut transversely into 5 slices of equal thickness followed by incubation in 1% triphenyltetrazolium chloride (TTC, Sigma)/PBS at 37°C for 8 minutes and then were fixed in 10% formalin–PBS for 24 hours. Fixed slices were then scanned, and ImageJ software (National Institute of Health, Bethesda, Maryland) was used to measure the infarct area. Infarct area was calculated as % non-TTC-stained area/total ventricular area.
Statistical Analysis
Data were analyzed, and the results were presented as mean ± standard deviation. Student’s t test or one-way analysis of variance was used for comparison of mean between the groups, and P < .05 was considered statistically significant.
Results
PT1 and 3HOI-BA-01 Protect Cardiomyocytes From OGD/R-Induced Apoptosis
Before testing whether PT1 and 3HOI-BA-01 are cardioprotective, we firstly tested whether these compounds are cytotoxic to cardiomyocytes under normal culture conditions. Apoptosis can be divided into early apoptosis and late apoptosis based on cell surface expression of phosphatidylserine and nuclear staining of PI. Early apoptotic cells are positively stained with annexin V while they are PI negative. Late apoptotic cells are both annexin V positive and PI positive. Necrotic cells are PI positive but annexin V negative. Treatment of mouse cardiomyocytes with PT1 and 3HOI-BA-01 at indicated doses for 24 hours did not significantly alter early or late apoptosis, suggesting that PT1 or 3HOI-BA-01 have little proapoptotic effect (Figure 1A and B). We then studied the effects of PT1 or 3HOI-BA-01 on OGD/R-induced cell death of cardiomyocytes. OGD/R significantly increased both early and late apoptosis, while PT1 and 3HOI-BA-01 profoundly inhibited apoptosis and necrosis, as well as promoted cell survival. Notably, PT1 and 3HOI-BA-01 together robustly inhibited OGD/R-induced apoptosis and maintained cell survival, suggesting a synergistic effect when they were used together (Figure 1C and D). Cell viability assay also confirmed the protective effects of PT1 and 3HOI-BA-01 (Figure 1E).

PT1 and 3H protect cardiomyocytes from OGD/R-induced apoptosis. A, Cardiomyocytes were treated with PT1 and 3H at indicated concentrations for 24 hours. Apoptosis was evaluated by flow cytometry. This is the representative of 3 independent experiments. B, Statistical analysis of cardiac myocyte apoptosis. C, Cardiomyocytes were subject to OGD/R followed by treatment with PT1 (20 µmol/L) and/or 3H (10 µmol/L). Cell apoptosis was evaluated by flow cytometry. This is the representative of 3 independent experiments. D, Statistical analysis of cardiac myocyte apoptosis. E, Cell viability assay using calcein-AM showed increased cell viability after PT1 and/or 3H treatment. n = 6 in each group. Ctrl, untreated control. V, vehicle. **P < .01; ***P < .001 compared with vehicle-treated OGD/R group. OGD/R indicates oxygen glucose deprivation/reoxygenation; 3H, 3HOI-BA-01.
PT1 and 3HOI-BA-01 Promote Autophagy in Cardiomyocytes
Autophagy has been shown to protect various cell types while in a state of nutrient insufficiency. 24,25 We expected that autophagy induced by PT1 and 3HOI-BA-01 would be cardioprotective after OGD/R. We first asked whether PT1 and 3HOI-BA-01 can induce autophagy in cardiomyocytes. Cardiomyocytes were treated with PT1 and/or 3HOI-BA-01 under normal culture conditions for 24 hours. OGD/R was conducted as a positive control for autophagy induction. Autophagic markers were detected by Western blotting after treatment (Figure 2A). We detected the conversion of LC3B to determine autophagy induction. LC3B is one of the 3 isoforms of LC3. Cleavage of LC3 at the carboxy terminus immediately following synthesis yields the cytosolic LC3-I form. During autophagy, LC3-I is converted to LC3-II through lipidation that allows for LC3 to become associated with autophagic vesicles. 26 PT1 or 3HOI-BA-01 significantly induced conversion of LC3B-I to LC3B-II, which is a marker for autophagosome formation. In addition, PT1 or 3HOI-BA-01 significantly upregulated Beclin-1 expression in cardiomyocytes. Beclin-1 is required for the initiation of autophagic vesicle formation. p62, which is degraded by autophagy, was decreased by PT1 or 3HOI-BA-01. OGD/R exposure induced similar but weaker changes of the above markers in comparison with PT1 or 3HOI-BA-01 treatment. Treatment with PT1 and 3HOI-BA-01 together induced the most significant changes in all 3 autophagic markers, suggesting combination of PT1 and 3HOI-BA-01 is the most potent autophagy activator. To confirm the effect of PT1 and 3HOI-BA-01 on autophagosome formation, immunofluorescence analysis of LC3B and LAMP-1 was performed. Anti-LC3B antibody staining displayed a homogenous distribution of LC3B, while LAMP-1 staining was relatively weak in the cytoplasm of vehicle-treated cells (Figure 2B). PT1 and/or 3HOI-BA-01 increased formation of autophagosomes, demonstrated by increased distribution of perinuclear LC3B spots, and colocalization of LC3B and LAMP-1-stained vesicles, which indicated autophagosome formation 27 (Figure 2B). However, LC3B staining cannot differentiate whether PT1 and 3HOI-BA-01 together induce higher autophagy than PT1 or 3HOI-BA-01 alone, since it is difficult to quantify autolysosome formation with staining. Western blotting was therefore used to determine the extent of autophagy. We tested whether PT1 and 3HOI-BA-01 together can promote autophagy in OGD/R-treated cardiomyocytes. Our finding was that PT1 plus 3HOI-BA-01 significantly enhanced LC3B-II level and decreased p62 level after cells were treated with OGD/R, indicating autophagy was substantially promoted in OGD/R-exposed cardiomyocytes. In addition, compared with OGD/R treatment alone, post-OGD/R treatment with PT1 plus 3HOI-BA-01 caused even more Beclin-1 and less p62 (Figure 2C). Taken together, our data indicated that both PT1 and 3HOI-BA-01 were able to induce autophagy.

PT1 and 3H activate autophagy in cardiomyocytes. A, Cardiomyocytes were treated with PT1 (20 µmol/L) and/or 3H (10 µmol/L) for 24 hours. Conversion of LC3B, protein levels of Beclin-1, and p62 were detected by Western blot. β-Actin was used as internal control. Left panel, Representative Western blot images of 4 independent experiments. Right panel, statistical analysis. B, Immunofluorescent analysis of LC3B and LAMP-1 in cardiomyocytes (200×). C, Cardiomyocytes were exposed to OGD/R followed by treatment with PT1 (20 µmol/L) and 3H (10 µmol/L). Conversion of LC3B and p62 level was detected by Western blot. Left panel, Representative Western blot images of 3 independent experiments. Right panel, statistical analysis. V, vehicle. Ctrl, untreated control. *P < .05; **P < .01; ***P < .001. LC3B indicates light chain 3 isoform B; LAMP-1, rat anti-lysosomal-associated membrane protein-1; OGD/R, oxygen glucose deprivation/reoxygenation; 3H, 3HOI-BA-01.
PT1 and 3HOI-BA-01 Protect Cardiomyocytes After OGD/R Through Induction of Autophagy
To confirm the role of autophagy in PT1 and 3HOI-BA-01-induced cardioprotection, we treated cardiomyocytes with PT1 and 3HOI-BA-01 in the presence or absence of chloroquine. Chloroquine inhibits autophagy by imparing protein degradation. Chloroquine significantly inhibited reduction of p62 induced by PT1 plus 3HOI-BA-01, suggesting autophagy was indeed blocked (Figure 3A). Since the degradation of LC3B-II was also blocked by chloroquine, the LC3B-II level in chloroquine-treated cells was increased (Figure 3A). Compared with vehicle-treated cells, chloroquine alone did not alter Beclin-1 expression (Figure 3A). The evaluation of cell death after OGD/R showed that the protective effect of PT1 and 3HOI-BA-01 was significantly inhibited when choloroquine was applied (Figure 3B and C). These data suggested that the cardioprotective efficacy of PT1 and 3HOI-BA-01 was indeed mediated by induction of autophagy.

Chloroquine abolished cardioprotective effects of combination of PT1 and 3H. A, Cardiomyocytes were exposed to OGD/R followed by treatment with PT1 (20 µmol/L) and 3H (10 µmol/L). Chloroquine inhibits autophagy induced by PT1 or 3H. Note that p62 level was decreased by PT1 or 3H, while chloroquine stabilized p62 level by inhibiting autophagy. Left panel, Representative Western blot images of 3 independent experiments. Right panel, statistical analysis. *P < .05; **P < .01; ***P < .001. B, Cardiomyocyte apoptosis was evaluated by flow cytometry after exposure to OGD/R. This is the representative of 5 independent experiments. C, Statistical analysis of cardiomyocyte apoptosis. n = 5 in each group. V, vehicle. Ch, chloroquine. *P < 0.05; **P < 0.01; ***P < 0.001 compared with the OGD/R + PT1 + 3H group. OGD/R indicates oxygen glucose deprivation/reoxygenation; 3H, 3HOI-BA-01.
PT1 and 3HOI-BA-01 Induce Autophagy Through Activating AMPK and Inhibiting mTORC1 Signaling in Cardiomyocytes
To confirm that AMPK and mTOR signaling are altered by PT1 and 3HOI-BA-01, we examined the activation status of these signal pathways in cardiomyocytes. Activating phosphorylation level of AMPKα was significantly higher in the presence of PT1 than in the nontreated group and 3HOI-BA-01 group (Figure 4A). ACC phosphorylation was also enhanced in the presence of PT1, indicating enhanced AMPK activity (Figure 4A). 3HOI-BA-01 slightly increased phosphorylation of AMPKα and ACC but the increase was not statistically significant (Figure 4A). Meanwhile, phosphorylation of p70S6 K and 4EBP1 was decreased by either PT1 or 3HOI-BA-01, suggesting mTORC1 signaling is inhibited after treatment (Figure 4B). PT1 plus 3HOI-BA-01 remarkably decreased phosphorylation of p70S6 K and 4EBP1 (Figure 4B).
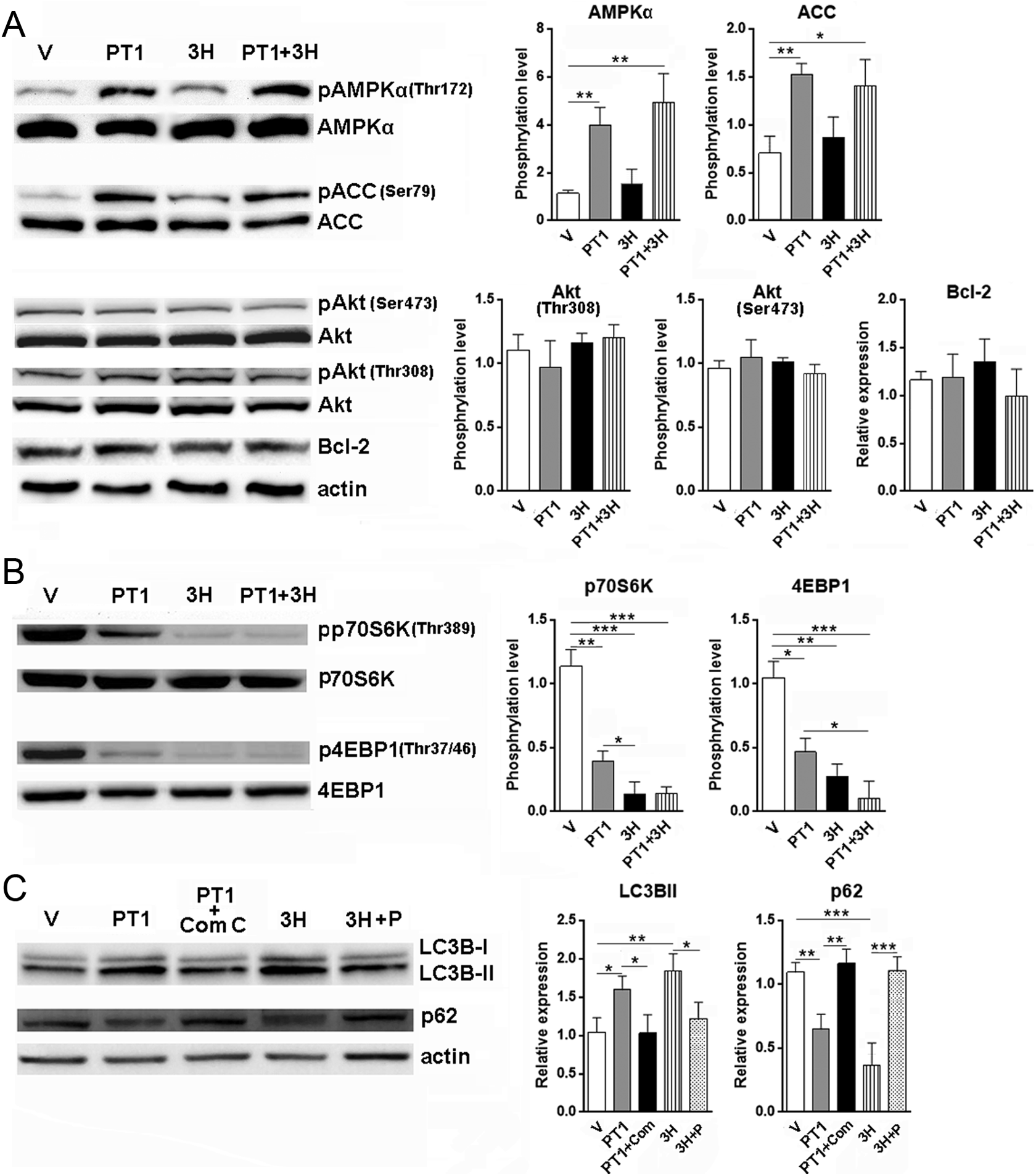
PT1 and 3H induce autophagy in cardiomyocytes via AMPK and mTOR signal pathway. A, Cardiomyocytes were treated with PT1 (20 µmol/L) and/or 3H (10 µmol/L) for 24 hours. AMPKα phosphorylation, ACC phosphorylation, Akt phosphorylation, and Bcl-2 expression were determined by Western blot. B, Phosphorylation of p70S6K and 4EBP1 was detected in cardiomyocytes treated with PT1 and/or 3H. C, Compound C and pervanadate were used to block AMPK and mTOR signaling in cardiomyocytes treated with PT1 or 3H. LC3B-II conversion and p62 level were determined by Western blot. n = 6 in each group. V, vehicle. Com C, compound C. P, pervanadate. *P < .05; **P < .01; ***P < .001. AMPK indicates 5′-adenosine monophosphate-activated protein kinase; mTOR, mammalian target of rapamycin; ACC, acetyl-coenzyme A carboxylase; Bcl-2, B-cell lymphoma 2; p70S6K, p70S6 kinase; 4EBP1, 4E-binding protein 1; LC3B, light chain 3 isoform B; 3H, 3HOI-BA-01.
To test whether other autophagy-related signal pathways are involved in PT1-and-3HOI-BA-01-induced autophagy, we examined the phosphorylation of Akt and the expression of Bcl-2 in cardiomyocytes. No alteration was found after treatment with PT1 or 3HOI-BA-01, suggesting neither agent impacted these signal pathways (Figure 4A).
Both AMPK and mTOR signaling are closely related to autophagy in many cell types including cardiomyocytes. 28 –30 To directly link AMPK and mTOR signaling to PT1-and-3HOI-BA-01-induced autophagy, we used a specific AMPK inhibitor, compound C to block AMPK signaling. We also used pervanadate, a protein tyrosine phosphatase (PTP) inhibitor, to activate the Akt-mTOR signal pathway. In the presence of compound C, PT1-induced LC3B-II conversion was significantly inhibited, and p62 level was stabilized (Figure 4C). This result suggests that AMPK signaling is involved in PT1-induced autophagy. Similar changes were observed after 3HOI-BA-01 treatment in the presence of pervanadate, suggesting mTOR signaling is a key player in 3HOI-BA-01-induced autophagy (Figure 4C).
PT1 and 3HOI-BA-01 Induce Autophagy in Mouse Hearts In Vivo
To test whether PT1 and 3HOI-BA-01 induce autophagy in cardiomyocytes in vivo, PT1 and/or 3HOI-BA-01 were intraperitoneally injected into mice. Then autophagic markers, AMPK, and mTOR signaling in hearts were determined by Western blot (Figure 5). Consistent with in vitro results, both PT1 and 3HOI-BA-01 increased LC3B-II conversion, while treatment with PT1 plus 3HOI-BA-01 induced the most robust conversion. PT1 promoted AMPKα phosphorylation while 3HOI-BA-01 reduced 4EBP1 phosphorylation, demonstrating their capability of activating AMPK signaling and inhibiting mTOR signaling, respectively. No significant change in Akt phosphorylation was found. Taken together, our data indicated that PT1 and 3HOI-BA-01 were able to induce autophagy in mouse hearts through regulating AMPK and mTOR pathways.
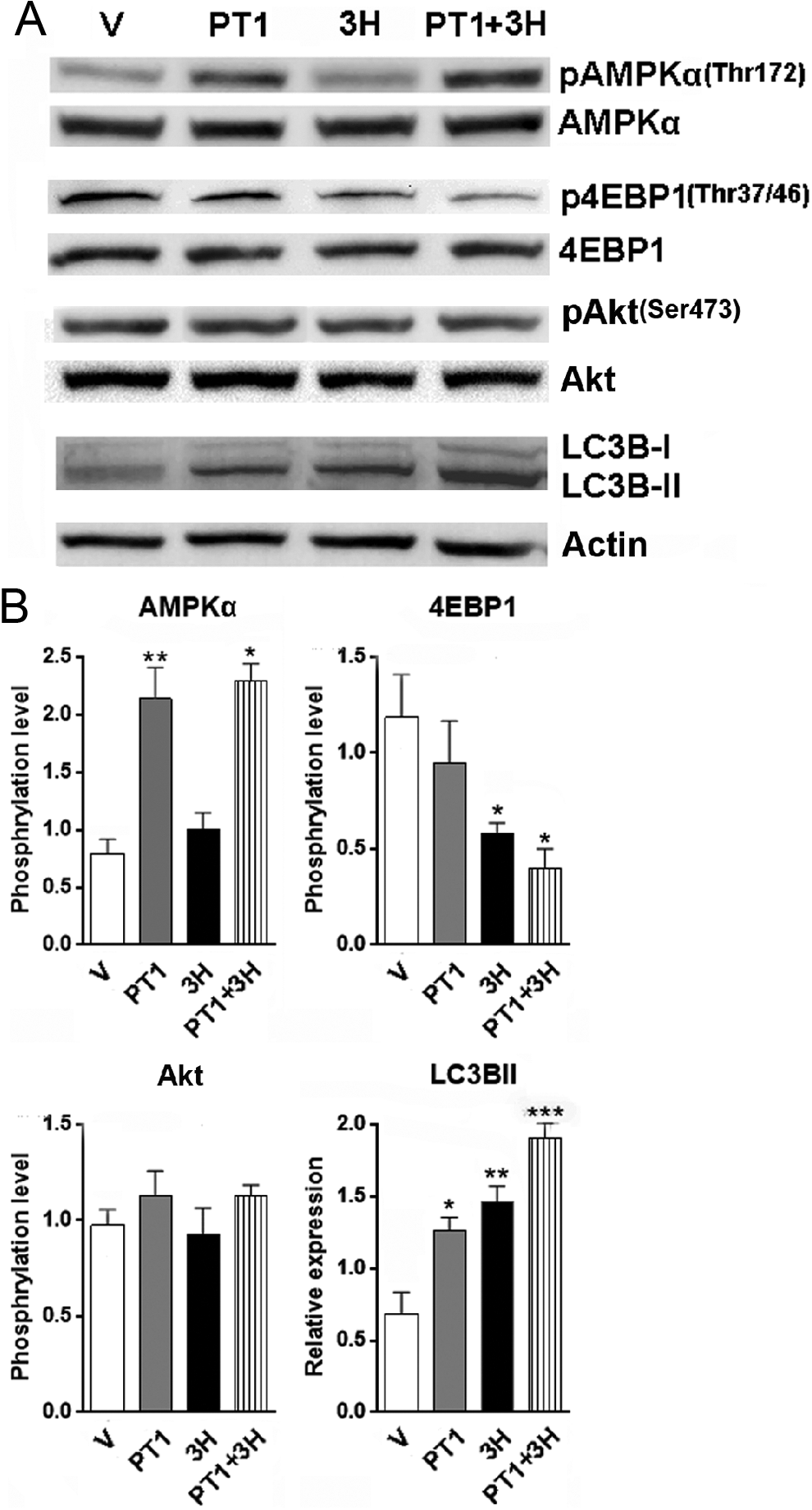
In vivo induction of autophagy in murine hearts by PT1 and/or 3H. PT1 and/or 3H was intraperitoneally injected into mice before AMPK signaling, mTOR signaling and LC3B conversion were determined by Western blot. A, Representative Western blot images of 5 independent experiments. B, Statistical analysis for Western blot. n = 5 in each group. V, vehicle. *P < .05; **P < .01. AMPK indicates 5′-adenosine monophosphate-activated protein kinase; mTOR, mammalian target of rapamycin; LC3B, light chain 3 isoform B; 3H, 3HOI-BA-01.
Treatment With PT1 Plus 3HOI-BA-01 Protects Mouse Hearts From I/R -Induced Damage
To explore the therapeutic application of PT1 and 3HOI-BA-01 in cardiac protection from I/R-induced myocardial damage, we applied a mouse myocardial I/R injury model and injected PT1 and 3HOI-BA-01 from the time point of reperfusion. We found that the infarct size was significantly reduced in the group receiving PT1 and 3HOI-BA-01 (Figure 6A). I/R itself promoted LC3B-II level and reduced p62 level, suggesting autophagy was induced in cardiomyocytes as a response to I/R. Furthermore, PT1 and 3HOI-BA-01 profoundly enhanced myocardial autophagy in comparison with vehicle control, as demonstrated by higher LC3B-II level and lower p62 level (Figure 6B and C). Our data suggest that combination treatment with PT1 and 3HOI-BA-01 could be a promising therapy for myocardial I/R.

Combination of PT1 and 3H reduced infarct size and induced autophagy in a murine myocardial ischemia/reperfusion model. A, PT1 and 3H were intraperitoneally injected into mice receiving myocardial ischemia/reperfusion. TTC staining was applied to analyze the infarct size in the hearts. Left panel, Representative TTC staining images. Right panel, statistical analysis for infarct size. B and C, Lysates of heart tissue were used to evaluate the induction of autophagy after ischemia/reperfusion by Western blot. n = 6 in each group. *P < .05; **P < .01. TTC indicates triphenyltetrazolium chloride; 3H, 3HOI-BA-01.
Discussion
Autophagy is a major catabolic pathway by which mammalian cells degrade and recycle macromolecules and organelles. Autophagy is crucial for removing protein aggregates and damaged/excess organelles to sustain intracellular homeostasis and to maintain the cell homeostasis. 31 In normal hearts, autophagy is kept at low levels under physiological conditions. However, the autophagic pathway is swiftly initiated and upregulated by environmental stress. 32 –34 Recent studies have revealed that the autophagosome number dramatically increases in the heart during/after ischemia/perfusion (I/R) in animal models. 35,36 Promoted autophagy has also been discovered in isolated and in vitro-cultured cardiomyocytes, in which exposure to hypoxia/reoxygenation caused increased number of autophagic vesicles. Autophagy-inducing signals during I/R include changed ATP levels and AMPK, hypoxia and Bnip3, calcium, reactive oxygen species and reactive N2 species, mitochondrial permeability transition pore opening, endoplasmic reticulum stress, and the unfolded protein response. The function of I/R-induced autophagy is still controversial, since many studies have demonstrated that autophagy plays a cardioprotective role in the heart following myocardial infarction. Other studies suggest autophagy is detrimental as a cause or contributor to cell death. 37,38 It is therefore speculated that the beneficial or detrimental role of autophagy is a consequence of balance depending on the extent of autophagy. The possible explanations for the cardioprotection by autophagy include ATP maintenance, mitophagy, protein clearance, and nutrient recycling. Thus, many studies are attempting to search for agents that can properly enhance autophagy during or after cardiac infarction.
Our study shows that PT1 and 3HOI-BA-01 can effectively protect in vitro-cultured cardiomyocytes after OGD/R. PT1 was identified and shown to possess a high potency toward AMPK, a specific AMPK agonist. 14 PT1 enhances AMPK activation in L6 myotubes in a time-dependent and dose-dependent manner. PT1 also lowers hepatic lipid content in a dose-dependent manner through AMPK activation in HepG2 cells, and this effect can be diminished by compound C. AMPK plays a key role as a master regulator of cellular energy homeostasis. This kinase is activated in response to stresses that deplete cellular ATP supplies such as low glucose, hypoxia, ischemia, and heat shock. AMPK activation positively regulates signaling pathways that replenish cellular ATP supplies. It also stimulates catabolic processes such as fatty acid oxidation and glycolysis via inhibition of ACC and activation of PFK2. 39 In addition, it has been shown that AMPK signaling positively regulates autophagy by activating Ulk1 through phosphorylation of Ser 317 and Ser 777, or indirectly by inhibiting mTOR signaling. 40,41 Our data indicate that PT1 enhances AMPK signaling and subsequently augments autophagy in cardiomyocytes. However, it is still not clear whether other mechanisms, such as AMPK-induced replenishment of cellular ATP, or downregulation of gluconeogenesis are involved in the protective effects. Further investigation will be needed to thoroughly elucidate the effects of PT1 on cardiomyocytes. In addition, since AMPK is a crucial mechanism by which cellular metabolism is regulated, it will be necessary to investigate whether short-term or long-term use of PT1 has impact on the metabolism of hearts and other organs or tissues.
3HOI-BA-01 is a newly discovered compound possessing in vitro and in vivo antitumor activity mediated through attenuation of mTOR signaling. 18 3HOI-BA-01 could potently suppress mTOR activity in a concentration-dependent manner. According to the molecular docking data, 3HOI-BA-01 formed 4 hydrogen bonds with the mTOR protein. One hydrogen bond involved the backbone atoms of the hinge residue Val2240 and the other 3 hydrogen bonds were formed with the side chain atoms of Lys2187, Tyr2225, and Asp2357, respectively. These results indicate that 3HOI-BA-01 may inhibit mTOR by directly binding with that protein. The mTOR signal pathway is the key regulator of autophagy. 42,43 High mTOR activity prevents Ulk1 activation by phosphorylating Ulk1 Ser 75728. 44 Ulk1 is one of the mammalian autophagy-initiating kinases. Ulk1 activation and association with other molecules such as Atg13 and FIP200 initiate autophagic membrane nucleation. Some other signal pathways, for example, PI-3K/Akt signaling and mitogen-activated protein kinase/Erk1/2 signaling, influence autophagy indirectly through regulating mTOR pathway. 45 Our study proves that 3HOI-BA-01 is protective for cardiomyocytes after OGD/R. Furthermore, PT1 also can inhibit mTOR signaling, probably due to the activation of AMPK. When PT1 and 3HOI-BA-01 are administered together, mTOR signaling is robustly inhibited and autophagy is highly promoted, compared with either agent alone. It is noteworthy that OGD/R itself also induces autophagy in cardiomyocytes, but the extent of induction is much less than that driven by PT1 and 3HOI-BA-01. Thus, we concluded that enhanced autophagy by PT1 and 3HOI-BA-01 is beneficial for cardiomyocyte survival. However, we cannot exclude the possibility that excessive induction of autophagy by abuse of these agents could cause severe myocardial damage. Appropriate dosages of PT1 and 3HOI-BA-01 are critical.
For potential clinical application of PT1 and 3HOI-BA-01, an important concern is their potential side effects. To our knowledge, neither agent has been subject to tests for any side effects so far. We used intraperitoneal injection because it is a relatively easy way to distribute the agents into blood circulation to access cardiomyoctes. Although we did not observe any significant abnormalities in the appearance and behavior of the mice tested, we cannot rule out the possibility that these agents at high blood concentrations could damage vital organs, especially liver, kidney, and lung. Hence, further research on the side effects of these 2 agents will give us more information on their safe medicinal use.
Conclusion
Taken together, our research demonstrated cardioprotective effects of PT1 and 3HOI-BA-01 after I/R injury exposure. Based on our discovery, a potentially novel therapeutic approach could be proposed for myocardial damage after I/R.
Footnotes
Authors’ Note
The authors Ling Huang and Kai Dai have contributed equally to this study. The work reported was done in Renmin Hospital of Wuhan University and The Central Hospital of Wuhan.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Sciences Foundation of China (No. 81300214).