
Abstract
Introduction:
Studies have shown that ticagrelor has a further adenosine-mediated mechanism of action in addition to its potent inhibition of the P2Y12 receptor, which may explain some of ticagrelor’s clinical characteristics. This study aimed to further characterize the adenosine pharmacology of ticagrelor, its major metabolites, and other P2Y12 receptor antagonists.
Methods:
Inhibition of nucleoside transporter-mediated [3H]adenosine uptake by ticagrelor, its major metabolites, and alternative P2Y12 antagonists was examined in recombinant Madin-Darby canine kidney (MDCK) cells. The pharmacology of ticagrelor and its major metabolites at adenosine A1, A2A, A2B, and A3 receptor subtypes was examined using in vitro radioligand binding and functional assays and ex vivo C-fiber experiments in rat and guinea pig vagus nerves.
Results:
Ticagrelor (and less effectively its metabolites) and the main cangrelor metabolite inhibited [3H]adenosine uptake in equilibrative nucleoside transporter (ENT) 1-expressing MDCK cells, whereas cangrelor and the active metabolites of prasugrel or clopidogrel had no effect. No significant inhibitory activity was observed in MDCK cells expressing ENT2 or concentrative nucleoside transporters 2/3. Ticagrelor demonstrated high affinity (inhibition constant [Ki] = 41 nmol/L) for ENT1. In adenosine receptor-binding experiments, ticagrelor and its major circulating metabolite, AR-C124910XX, had low affinity (Ki > 6 µmol/L) for each of the adenosine A1, A2A, and A2B receptors, whereas ticagrelor had a submicromolar (Ki = 190 nmol/L) affinity for the adenosine A3 receptor. However, in functional assays, at high concentrations (10 µmol/L) ticagrelor only partially inhibited 3 mmol/L adenosine-induced depolarizations in the guinea pig and rat vagus nerve preparations (by 35% and 49%, respectively).
Conclusions:
Ticagrelor inhibits cellular adenosine uptake selectively via ENT1 inhibition at concentrations of clinical relevance. However, the low-binding affinity and functional inhibition of adenosine receptors observed with ticagrelor or its metabolites indicate that they possess a negligible adenosine-like activity at clinically relevant concentrations.
Introduction
Ticagrelor is a direct-acting, reversibly binding oral P2Y12 antagonist 1,2 approved for treatment of patients with acute coronary syndromes (ACSs). The antiplatelet and clinical activity of ticagrelor is predominantly mediated through its potent and reversible inhibition of the platelet P2Y12 receptor. 2,3 In the phase III PLATelet inhibition and patient Outcomes (PLATO) trial (n = 18 624 patients with ACS), ticagrelor reduced the rate of the composite end point of myocardial infarction/stroke/death from vascular causes without an increase in overall major bleeding compared with clopidogrel. Moreover, all-cause mortality was also reduced in patients receiving ticagrelor compared with clopidogrel, 4 an effect not observed with other contemporary antiplatelet agents. Furthermore, during the clinical development program of ticagrelor, dyspnoea and ventricular pauses were observed in some patients. 3 –5 These observations are suggestive of an additional mechanism of action for ticagrelor.
Several other findings support such an additional mode of action via adenosine. For example, ticagrelor augmented both endogenous and exogenous adenosine-induced coronary blood flow in a canine model 6 and augmented exogenous adenosine-induced coronary blood flow increases and dyspnoea in healthy volunteers. 7 In vitro, ticagrelor has been shown to inhibit adenosine uptake into human erythrocytes as well as other human, rat, and canine cell lines 6 ; ticagrelor also inhibited ex vivo human platelet aggregation in whole blood via increases in adenosine levels. 8
Several clinical studies have examined the cardiovascular and respiratory effects of adenosine. 9 –12 Adenosine acting via A1, A2A, A2B, and A3 receptors may have beneficial effects in patients with ACS, including cardioprotection, vasodilatation, inflammatory regulation, and platelet function inhibition. 11 Adenosine may also mediate bradycardia through direct effects on the sinoatrial and atrioventricular nodes 10 and tachycardia via increased cardiac sympathetic drive and decreased vagal tone. 9,10 In addition, adenosine has been shown to cause dyspnoea in healthy volunteers, potentially through direct stimulation of pulmonary C-fiber endings, 12 a mechanism supported by in vivo data in rats 13 and guinea pigs. 14
Although ticagrelor is neither an adenosine analog, nor is it metabolized into adenosine, 15 it is important to characterize the pharmacology of ticagrelor and its metabolites toward adenosine transporters and receptors and compare it with that of other known P2Y12 antagonists. Adenosine uptake is mediated by members of the sodium-dependent concentrative nucleoside transporter (CNT) 2/3 family 16 and sodium-independent equilibrative nucleoside transporter (ENT) 1/2 family. 17 Identification of the transporter that ticagrelor inhibits to reduce adenosine uptake may also explain ticagrelor’s adenosine-mediated mechanism of action.
Hence, the objective of the present in vitro and ex vivo studies was to further characterize ticagrelor’s pharmacology with respect to adenosine. The effect of ticagrelor on specific adenosine transporters was examined using recombinant cell systems. Second, receptor–ligand binding and functional assays were performed, as a direct effect of ticagrelor and its metabolites at adenosine receptors has not been ruled out. Finally, as dyspnoea could result from activation of pulmonary C-fibers, the effects of adenosine and ticagrelor were assessed in ex vivo guinea pig and rat C-fiber preparations.
Materials and Methods
Materials
Cell culture media were sourced from Invitrogen (Paisley, United Kingdom). Transfection reagents were purchased from Life Technologies. [3H]S-(4-Nitrobenzyl)-6-thioinosine ([3H]NBTI; specific activity 8.4 Ci/mmol, 11.9 µmol/L) and [2,8 3H]adenosine (specific activity 40 Ci/mmol, 25 µmol/L) were purchased from ARC Inc (Saint Louis, Missouri). All other reagents and chemicals were purchased from Sigma (Dorset, UK) unless specified. Ticagrelor, AR-C124910XX (main circulating metabolite of ticagrelor and active vs P2Y12), AR-C133913XX (main urinary metabolite of ticagrelor and inactive vs P2Y12), 15 and elinogrel, clopidogrel active metabolite, prasugrel active metabolite and cangrelor main metabolite were synthesized in house (AstraZeneca, Mölndal, Sweden).
Cell Lines for Adenosine Uptake Experiments
Plasmids encoding human ENTs (hENTs) and CNTs and green fluorescent protein (protein IDs ENT1 [M1-V456], ENT2 [M1-L456], CNT1 [M1-Q649], CNT2 [M1-A658], CNT3 [M1F691]) were synthesized and inserted into pcDNA3.1 by GeneArt gene synthesis service. Transient transfection of Madin-Darby canine kidney (MDCK) cells with the constructs was performed using Lipofectamine 2000. A control cell line was created by transient transfection of MDCK cells with an empty pcDNA3.1 expression construct (MOCK) using MaxCyte electroporation equipment. Transiently transfected cells were maintained in growth medium containing G418 (400 µg/mL).
Cell membrane preparations for competitive binding experiments with NBTI required the dissociation of the transfected MDCK cells with accutase, resuspension in Dulbecco phosphate-buffered saline (DPBS) containing Ca2+ and Mg2+ and protease inhibitor cocktail, and disruption via sonication (Soniprep 150, MSE (UK) Ltd, London, UK). Subsequent centrifugation at 3000g for 3 minutes at 4°C and further centrifugation of the homogenate at 30 000g for 30 minutes at 4°C preceded resuspension of the crude membranes in DPBS (pH7.4) without protease inhibitor.
Examination of [3H]Adenosine Uptake by ENTs and CNTs
The effect of the following test compounds (Figure 1) on [3H]adenosine uptake in MDCK cell lines expressing specific ENTs and CNTs was examined: ticagrelor, AR-C124910XX, AR-C133913XX, cangrelor, cangrelor main metabolite, elinogrel, clopidogrel active metabolite, prasugrel active metabolite, dipyridamole (adenosine transport inhibitor), and NBTI (adenosine transport inhibitor); unless stated all compounds were examined at 0.1, 1, 10, and 100 nmol/L, and 1, 10, 30, and 100 µmol/L. Transfected cells were seeded into 96-well scintiplates (PerkinElmer ScintiPlate-96 TC, PerkinElmer, Cambridge, UK; ENT1/2-transfected cells: 2 × 104 cells/well; CNT2/3-transfected cells 3 × 104 cells/well). Equilibrative nucleoside transporter 1/2 cells after 24 hours or CNT2/3 cells after 72 hours were exposed to either test compound or control in the presence of 90 nmol/L [3H]adenosine substrate in sodium-free (ENT1/2) 6 or sodium-containing buffer (CNT2/3). The reaction was stopped after 30 minutes with sodium-free buffer containing 100 µmol/L dipyridamole (ENT1/2) or 5 mmol/L cold adenosine (CNT2/3). [3H]Adenosine uptake was measured using proximity scintillography and normalized for total protein concentration. All assays were performed on ≥3 occasions except the control MOCK experiments for the CNT2/3 studies where N = 1.
Structures of compounds used in the adenosine uptake studies: (A) ticagrelor, (B) AR-C124910XX, (C) AR-C133913XX, (D) cangrelor, (E) cangrelor main metabolite, (F) elinogrel, (G) clopidogrel active metabolite, (H) dipyridamole, and (I) prasugrel active metabolite.
Examination of the Dissociation of [3H]NBTI by Ticagrelor and Its Metabolites and Other P2Y12 Receptor Antagonists
Competitive binding studies were performed to examine whether ticagrelor was interacting directly with ENT1 to inhibit [3H]adenosine uptake. Compounds that demonstrated inhibition of adenosine uptake by ENT1 were assessed for their ability to displace [3H]NBTI in MDCK cells expressing hENT1 and MDCK-MOCK. Membrane homogenates (final concentration 50 µg/mL) were incubated in a total 1.8 mL of DPBS (with Ca2+ and Mg2+) with each compound and 2 nmol/L [3H]NBTI for 60 minutes at room temperature. Incubation was terminated by rapid filtration through Whatman 350 GF/B filter plates, and the remaining radioactivity was measured by liquid scintillation (Ultima GOLD F, Perkin Elmer) counting. Nonspecific binding was defined in the presence of 2 µmol/L unlabeled NBTI. Specific binding was calculated by subtracting the nonspecific binding from the total binding. All assays were performed on ≥3 occasions.
The Role of Ticagrelor and Its Metabolites at Adenosine Receptor Subtypes in Recombinant Cells
The activity of ticagrelor, AR-C124910XX, and AR-C133913XX at adenosine receptor subtypes was investigated using standard radioligand binding and functional assays in Chinese hamster ovary cells by Ricerca BioSciences LLC (Taipei, Taiwan), Cerep (Celle L’evescault, France), and Calliper Life Sciences (Hanover, Maryland). To further investigate the functional consequence of ticagrelor interaction with the A2A receptor, a single concentration of either the reference agonist, adenosine-5′N-ethylcarboxamide (NECA; 10 nmol/L), or ticagrelor (1 μmol/L) was used to stimulate the A2A receptor and a range of 10 concentrations of the A2A selective reference antagonist, 8-(3-chlorostyryl) caffeine (CSC), were used to inhibit these responses in human embryonic kidney cells. All experiments were performed on ≥3 occasions.
The Role of Ticagrelor at Adenosine Receptor Subtypes in an Ex-Vivo Vagus Preparation
As adenosine may mediate dyspnoea by its reported action on pulmonary C-fibers, the effects of adenosine and ticagrelor were assessed in ex-vivo guinea pig and rat C-fiber preparations. Male Dunkin Harley guinea pigs (Harlan, United Kingdom) and male Wistar rats (Harlan) were acclimatized for 7 days prior to sacrifice via cervical dislocation or pentobarbitone overdose, respectively. Vagus nerves were removed as described previously 13 and placed in Krebs solution and bubbled with 95% O2/5% CO2. Nerves were cleared of connective tissue and desheathed; nerve trunks remained in oxygenated Krebs solution throughout. The methodology for the depolarization recording from the vagus was as reported previously. 18,19
Drugs were applied into the perfusion solution of the first channel only and the response recorded via a chart recorder. Responses to 100 nmol/L to 30 mmol/L adenosine were determined in the guinea pig vagus (n ≥ 6), and responses to 1 to 10 mmol/L adenosine were determined in the rat vagus (n = 2). Control responses to capsaicin (1 µmol/L, perfused for 2 minutes) were obtained at the beginning and end of each experiment. Selective antagonists for the adenosine A1 (DPCPX), A2A (SCH58261), A2B (MRS1706), and A3 (MRS1220) receptors were used to further characterize the guinea pig (n = 6) and rat nerves (n = 4) using the following sequence: (1) response to 1 µmol/L capsaicin perfusion for 2 minutes, (2) 2 reproducible responses to 3 mmol/L adenosine perfusion for 2 minutes, (3) 10-minute incubation with 3 µmol/L selective antagonist, (4) 2-minute stimulation with 3 mmol/L adenosine in presence of antagonist, (5) 2-minute stimulation with 3 mmol/L adenosine, (6) final response to 1 µmol/L capsaicin perfusion for 2 minutes, and (7) washout until responses returned to baseline.
The above-mentioned protocol was used to test the effects of ticagrelor on 3 mmol/L adenosine-induced depolarization of guinea pig (ticagrelor doses 0.3, 1, 3, and 10 µmol/L; n = 4) and rat (ticagrelor dose 10 µmol/L; n = 4) vagus nerves; direct activation of vagus C-fibers by ticagrelor was carefully monitored during the 10-minute incubation period (n = 4, both models). The effect of an adenosine deaminase inhibitor erythro-9-(2-hydroxy-3-nonyl) adenine hydrochloride (EHNA; n = 2) and an adenosine transport inhibitor (dipyridamole; n = 4) on the adenosine dose–response curve was also investigated in the guinea pig nerve preparations. Adenosine solutions, vehicle controls (water for EHNA; 0.1% dimethyl sulfoxide for dipyridamole), 10 µmol/L EHNA, or 1 or 10 µmol/L dipyridamole were added via the Krebs solution.
Data Analysis and Statistics
Prism 5.0 software (Graphpad Software Inc, San Diego, California) was used to calculate the micromolar concentration of test compound producing 50% inhibition (IC50) ± standard error of the mean (SEM) using nonlinear regression (log(inhibitor) vs response). Inhibition constant (Ki) values were derived from IC50 values based on the equation of Cheng and Prusoff. 20 The effect of each antagonist, or ticagrelor, on adenosine-induced depolarization of C-fiber preparations was analyzed using a paired student t test comparing adenosine responses before and after incubation with antagonist or ticagrelor. Differences were deemed statistically significant when P < 0.05.
Results
Effect of Ticagrelor and Its Metabolites and Other P2Y12 Receptor Antagonists on [3H]adenosine Uptake in Transfected MDCK Cells
[3H]Adenosine uptake was similar (1-4 pmol/µg/min) in cells expressing ENT1, ENT2, CNT2, or CNT3 (Figure 2). In cells expressing CNT-2/3, [3H]adenosine uptake was shown to be sodium dependent (Figure 2). [3H]Adenosine transport was negligible in cells expressing CNT1 (data not shown), and subsequent screening against this transporter was not performed. Minimal [3H]adenosine uptake was observed in control MOCK-transfected cells (Figure 2). Dipyridamole, a known inhibitor of the nucleoside transporters, inhibited [3H]adenosine uptake in each cell line (data not shown).

Rate of uptake of [3H]adenosine (90 nmol/L) in MDCK cells transiently transfected with (A) ENT1 or ENT2 (sodium-free buffer only) or (B) CNT2 or CNT3; in each series of experiments nontransporting (MOCK) constructs were assessed for control purposes. Data shown are mean of ≥3 experiments ± SEM (except MOCK in (B) where n = 1). CNT indicates concentrative nucleoside transporter; ENT, equilibrative nucleoside transporter; MDCK, Madin-Darby canine kidney; SEM, standard error of the mean.
In cells expressing ENT1, ticagrelor inhibited [3H]adenosine uptake with an IC50 of 260 nmol/L. AR-C124910XX, AR-C133913XX, and the metabolite of cangrelor also inhibited adenosine uptake (Table 1). No other P2Y12 antagonists inhibited [3H]adenosine uptake at an IC50 <10 µmol/L (Table 1). The order of potency with respect to this uptake was ticagrelor > cangrelor main metabolite > AR-C124910XX > AR-C133913XX. However, ticagrelor was about 35-fold less potent than that of dipyridamole (IC50 7.4 nmol/L).
Effect of P2Y12 Receptor Antagonists and Dipyridamole on Adenosine Transport as Measured by Inhibition of [3H]Adenosine Uptake in MDCK Cell Lines Expressing Specific ENTs and CNTs.a

Abbreviations: CNTs; concentrative nucleoside transporters; ENTs, equilibrative nucleoside transporters; MDCK, Madin-Darby canine kidney; SEM, standard error of the mean; pIC50, negative logarithm of the IC50; IC50, the concentration causing 50% inhibition of signal.
a Data are mean of ≥3 experiments (pIC50 ± SEM); mean IC50 (µmol/L) is shown in parentheses.
In cells expressing ENT2, CNT2, or CNT3, [3H]adenosine uptake was not inhibited by ticagrelor or any P2Y12 antagonist, with all IC50 values less than 10 µmol/L (Table 1).
To confirm that the observed inhibition of [3H]adenosine uptake was mediated through inhibition of ENT1, we tested whether each compound could cause dissociation of the ENT1 ligand [3H]NBTI from ENT1 (Table 2). Ticagrelor demonstrated a high affinity for ENT1 (Ki = 41 nmol/L), which was approximately 15-fold less than that of dipyridamole, 2.6 nmol/L (Table 2). AR-C124910XX and AR-C133913XX also displaced [3H]NBTI from ENT1 (Table 2 and Figure 3) but with lower affinities (0.3 µmol/L and 2.3 µmol/L, respectively). The dissociation constant (Kd) of [3H]NBTI was 0.29 nmol/L.
Effect of P2Y12 Receptor Antagonists and Dipyridamole on the Inhibition of [3H]NBTI Binding to ENT1 in MDCK Cells Expressing ENT1.a
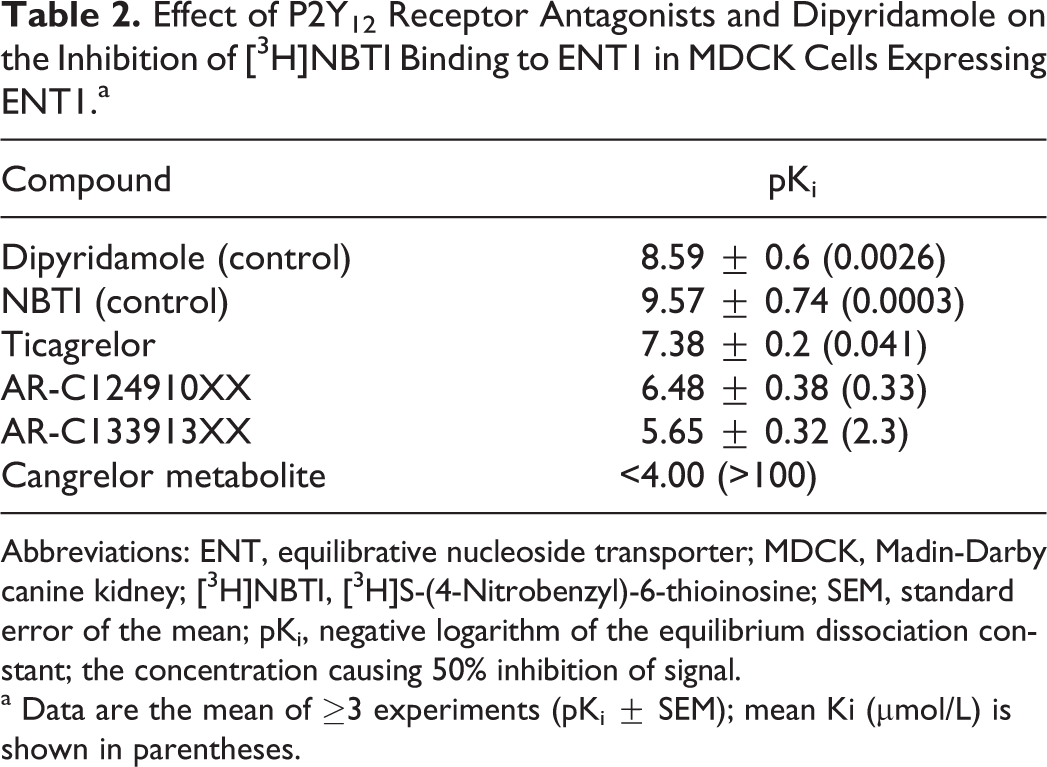
Abbreviations: ENT, equilibrative nucleoside transporter; MDCK, Madin-Darby canine kidney; [3H]NBTI, [3H]S-(4-Nitrobenzyl)-6-thioinosine; SEM, standard error of the mean; pKi, negative logarithm of the equilibrium dissociation constant; the concentration causing 50% inhibition of signal.
a Data are the mean of ≥3 experiments (pKi ± SEM); mean Ki (µmol/L) is shown in parentheses.
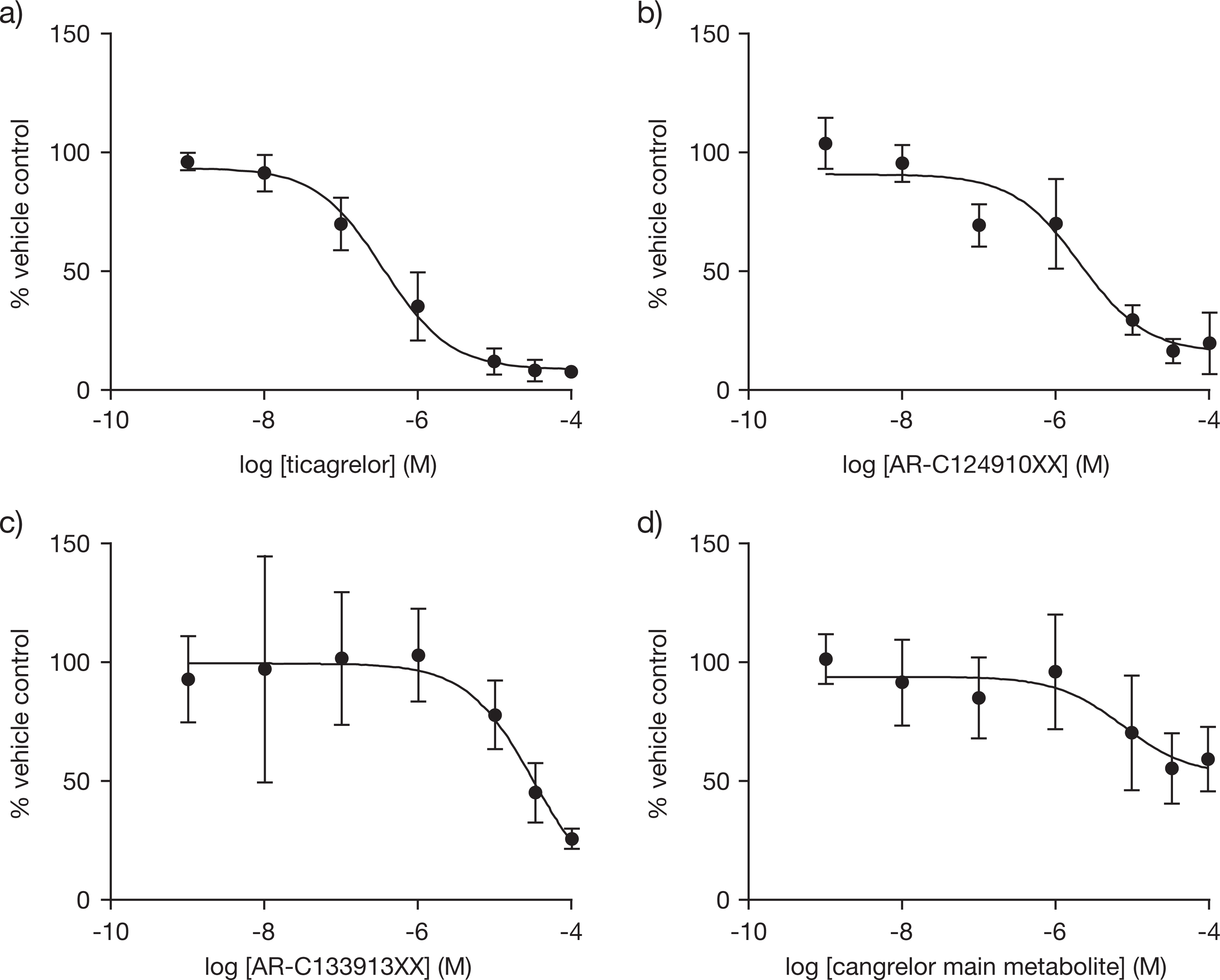
Inhibition of [3H]NBTI binding in MDCK-ENT1 cells by (A) ticagrelor, (B) AR C124910XX, (C) AR-C133913XX, and (D) cangrelor main metabolite. Data shown are mean of 4 experiments ± SEM. [3H]NBTI indicates [3H]S-(4-nitrobenzyl)-6-thioinosine; MDCK, Madin-Darby canine kidney; ENT, equilibrative nucleoside transporter; SEM, standard error of the mean.
The Role of Ticagrelor at Adenosine Receptor Subtypes in Recombinant Cells
In radioligand-binding experiments, ticagrelor had low affinity (Ki > 6 µmol/L) for the A1, A2A, and A2B receptors (Table 3) and a submicromolar affinity for the adenosine A3 receptor (Ki = 0.19 µmol/L). In functional assays, ticagrelor inhibited the action of the reference agonists at the A1, A2B, and A3 receptors with IC50 values of 22 µmol/L, 23 µmol/L, and 6 µmol/L, respectively; AR C124910XX inhibited these receptor subtypes with a similar potency to ticagrelor, whereas the potency of AR-C133913XX was generally lower (Table 3).
Effect of Ticagrelor and its Metabolites at Adenosine Receptor Subtypes in CHO Cell Lines.a

Abbreviations: CHO, Chinese hamster ovary; SEM, standard error of the mean; pKi, logarithm of inhibition constant; the concentration causing 50% inhibition of signal; EC50, the concentration causing 50% maximum effect; negative logarithm of the IC50; pEC50, negative logarithm of the EC50.
a Data are the mean of ≥3 experiments (mean pKi, pIC50, and pEC50, each ± SEM); mean Ki, IC50, and EC50 (µmol/L) are shown in parentheses.
The concentration of ticagrelor causing 50% maximal effect (EC50) at the A2A subtype was 680 nmol/L (Table 3). To confirm that the agonist effect seen in the functional assay was due to interaction with the A2A receptor, a selective antagonist, CSC, was used to demonstrate that the signal caused by stimulation with NECA (reference agonist) or ticagrelor could be inhibited. Addition of CSC inhibited 10 nmol/L NECA- and 1 µmol/L ticagrelor-induced stimulation of the adenosine A2A receptor (Figure 4A and B).

Inhibition of (A) 1 µmol/L ticagrelor-induced and (B) 10 nmol/L NECA-induced stimulation of the adenosine A2A receptor by the selective antagonist CSC in HEK cells. Data shown are mean of ≥3 experiments ± SEM. CSC indicates 8-(3-chlorostyryl) caffeine; NECA, 5′N-ethylcarboxamide; HEK, human embryonic kidney; SEM, standard error of the mean.
The Role of Ticagrelor at Adenosine Receptor Subtypes in an Ex Vivo Vagus Preparation
Perfusion of adenosine onto guinea pig (n ≥ 6; Figure 5A) and rat (n = 2; Figure 5B) vagus nerve preparations resulted in reproducible depolarizations in a dose-dependent manner, confirming the activity of adenosine in this model; a maximum response was observed with a 10 mmol/L dose. Addition of 10 μmol/L EHNA (an inhibitor of adenosine deaminase) did not appear to change the magnitude of depolarization induced by 1, 3, or 10 mmol/L of adenosine in the guinea pig, relative to vehicle (n = 2; data not shown). Similarly, addition of 1 μmol/L or 10 μmol/L dipyridamole (an adenosine transport inhibitor) did not appear to change the magnitude of adenosine-induced depolarizations (0.3-3 mmol/L) in the guinea pig vagus relative to vehicle (n = 4; data not shown). A1, A2B, and A3 adenosine receptor antagonists (3 µmol/L DPCPX, 3 µmol/L MRS1706, and 3 µmol/L MRS1220, respectively) inhibited 3 mmol/L adenosine-induced depolarizations in guinea pig (n = 6; Figure 5C) and rat (n = 4; Figure 5D) vagus nerve preparations.
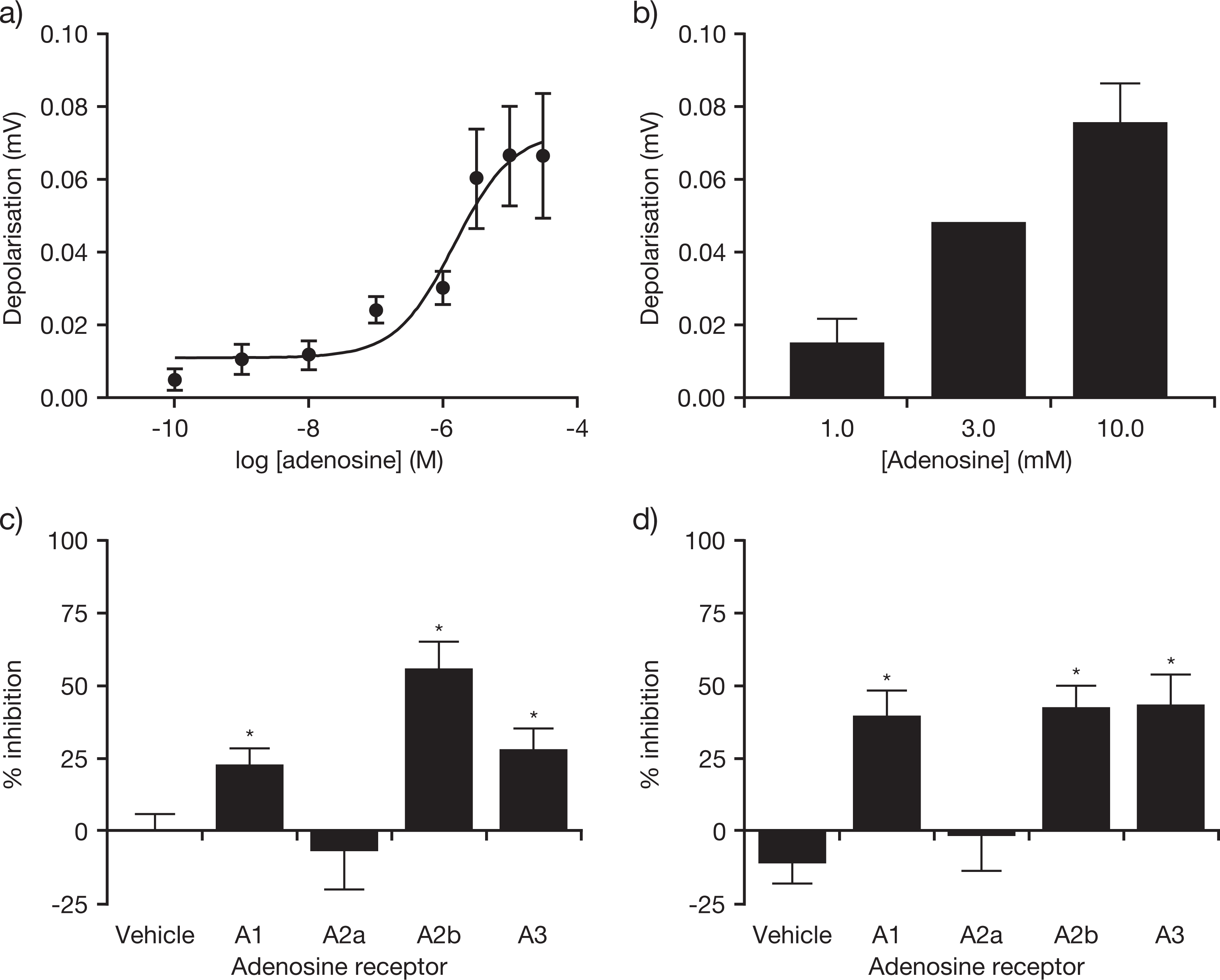
Effect of adenosine on (A) guinea pig and (B) rat vagus nerve preparations. Data shown are mean ± SEM; n ≥ 6 and n = 2 at each adenosine concentration in guinea pig and rat, respectively. The effect of the selective adenosine A1, A2A, A2B, and A3 receptor antagonists on adenosine-mediated depolarizations is shown in (C) guinea pig and (D) rat preparations. Data shown are mean ± SEM; n = 4 for each experiment; *P < .05 versus vehicle. SEM indicates standard error of the mean.
Addition of ticagrelor (10 µmol/L) partially inhibited 3 mmol/L adenosine-induced depolarizations in both the guinea pig (n = 4; Figure 6A) and the rat (n = 4; Figure 6B) vagus nerve preparations by 35% and 49%, respectively. Small depolarizations were also observed in response to 10 µmol/L ticagrelor in 3 of 6 guinea pig vagus nerve preparations (0.02 ± 0.01 mV) and 3 of 4 rat vagus nerve preparations (0.03 ± 0.01 mV).
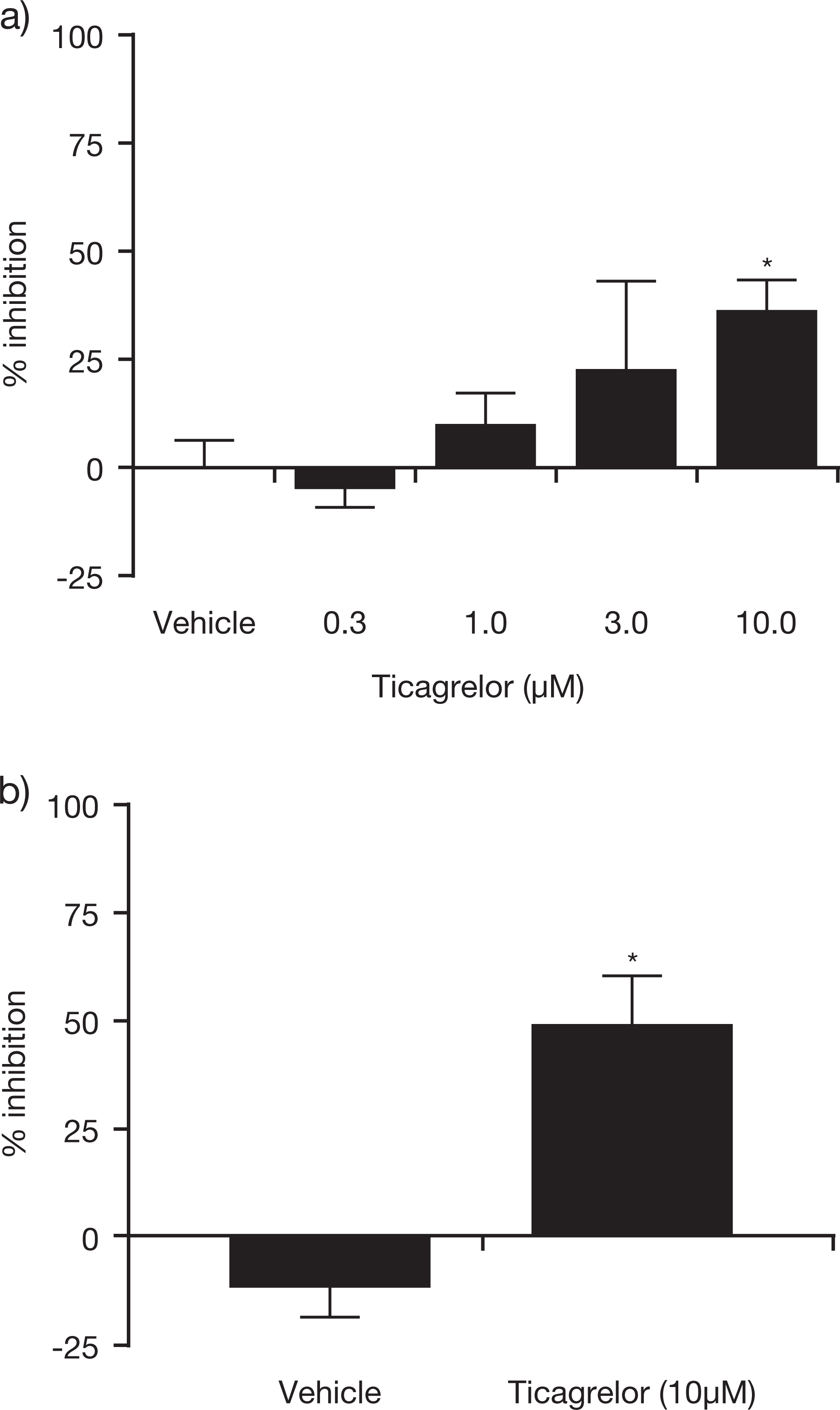
Effect of ticagrelor on adenosine-induced (3 mmol/L) depolarizations in (A) guinea pig and (B) rat vagus preparations. Data shown are mean ± SEM; n = 4 for each experiment; *P < .05 versus vehicle. SEM indicates standard error of the mean.
Discussion
The present in vitro and ex vivo studies were performed to further characterize the pharmacology of ticagrelor with respect to its adenosine-mediated effects. Our findings confirm that ticagrelor inhibits adenosine uptake via the ENT1 transporter and demonstrate that ticagrelor is unlikely to act directly at adenosine receptors to a clinically relevant extent. These data are in line with, and provide a mechanism for, the adenosine-mediated activity of ticagrelor as shown in previous studies. 6 –8
Ticagrelor inhibited adenosine uptake in MDCK cells expressing ENT1, with a potency (IC50) approximately 35-fold less than that of dipyridamole (a known inhibitor of ENT1). 21 These findings are generally consistent with those from a previous study in human breast carcinoma cells, where the potency of ticagrelor with respect to inhibition of adenosine uptake was approximately 10-fold less than that of dipyridamole. 6 Further, we have previously demonstrated that ticagrelor can augment adenosine-induced platelet inhibition by prolonging the half-life of adenosine added to whole blood. Via this mechanism, ticagrelor in effect increases the concentration of adenosine, which can subsequently interact via the platelet adenosine A2A receptor; the potency (EC50) of ticagrelor for this effect was approximately 5 µmol/L. 8 Here, we have also demonstrated that ticagrelor can displace [3H]NBTI from ENT1 in MDCK cells confirming that ticagrelor binds to ENT1 directly to inhibit adenosine uptake. The affinity of ticagrelor for ENT1 was 41 nmol/L, and the dissociation constant (Kd) of [3H]NBTI in this system was similar (0.29 nmol/L) to that reported previously. 17
Ticagrelor demonstrated low affinity (ie, Ki > 6 µmol/L) for the adenosine A1, A2A, and A2B receptor subtypes and a submicromolar affinity for the A3 subtype. Generally, we also showed low affinity interactions of the major circulating and urine metabolites of ticagrelor (AR-C124910XX and AR-C133913XX, respectively) with the A1, A2A, and A2B receptor subtypes. These findings were confirmed for ticagrelor in a different model; ex vivo guinea pig and rat C-fiber preparations. As expected, and in line with findings from earlier studies, 13,14 adenosine depolarized vagal C-fibers (guinea pig and rat) via the A1, A2B, and A3 receptors. Ticagrelor inhibited adenosine-mediated depolarization of vagal C-fibers at high concentrations only. Small depolarizations were also observed in response to 10 µmol/L ticagrelor alone. It is unlikely that A1 and A3 receptor subtypes were responsible for this effect, as the functional studies suggested that ticagrelor acts as a weak antagonist at these receptors. The results indicate that the A2A receptor was not involved in vagus nerve depolarization, and ticagrelor may possibly be acting as a partial agonist at the A2B receptor. To examine this theory, antagonism of the response to ticagrelor in the guinea pig vagus was tested with the A2B inhibitor, MRS1706. However, due to the lack of a reproducible response to ticagrelor, these experiments were not successful. A potential explanation for the variable effects of ticagrelor observed in the receptor experiments reported herein is that any effect for compounds with a low affinity for the receptor may be overshadowed by the inherent variability in signal in such experiments. Therefore, we conclude that it is unlikely that ticagrelor or its metabolite will induce a clinical response via specific adenosine receptor interactions.
A limitation in the interpretation of these findings is that low species homology between rat and human receptors 22 and prior knowledge that the xanthine class of adenosine receptor ligands displays different affinities between species (see Jacobson for review) 23 suggest that extrapolation between rat or guinea-pig models and humans must be made with caution. Indeed, it is not possible to directly extrapolate findings from the in vitro preparation studies to human pharmacology. For example, the extent of protein binding to ticagrelor is not compensated for in these in vitro experiments. In patients with ACS who received ticagrelor (90 mg twice daily) for 4 weeks, the steady state mean maximum plasma concentration was 1.5 µmol/L. 24 However, ticagrelor and its major circulating metabolite are extensively plasma protein bound in vivo (>99.7%), 25 meaning that the unbound concentration is in the low nanomolar range. In comparable in vitro experiments where plasma protein binding is also negligible, the Ki for ticagrelor versus 2MeS-ADP on the P2Y12 receptor is approximately 4 nmol/L. 2
Based on the affinity of ticagrelor for the ENT1 transporter (Ki = 41 nmol/L) and the strong plasma protein binding, we would therefore anticipate that at clinical doses, ticagrelor is likely to only partially inhibit ENT1. However, such mild inhibition is expected to be sufficient to increase local concentrations of adenosine in the circulation; we have previously demonstrated that the addition of 1 µmol/L ticagrelor to whole blood in vitro is sufficient to significantly conserve extracellular concentrations of adenosine, which were 3.4-fold higher in the presence of ticagrelor relative to vehicle only. 8 Given that adenosine is a direct-acting agonist at its receptors, any local elevation would be expected to increase receptor signaling and the resulting response. Indeed, ticagrelor (180 mg) augmented adenosine-induced coronary blood flow increases in healthy volunteers, an effect that correlated with plasma exposure and was more evident at plasma concentrations greater than 1 µmol/L. 7
Based on the results of the receptor binding studies, we conclude that a direct interaction of ticagrelor with adenosine receptors is unlikely to be of clinical relevance. The affinity of ticagrelor for the adenosine A3 receptor, the subtype with the highest affinity interaction observed with ticagrelor (Ki = 190 nmol/L), is about 5-fold lower than the affinity of ticagrelor for the ENT1 transporter (Ki = 41 nmol/L). A lack of clinically relevant effect for ticagrelor at the adenosine receptors is supported by data from Wittfeldt et al 7 who showed that the effect of ticagrelor on coronary blood flow velocity correlated with the plasma concentration of ticagrelor only in the presence of adenosine. Indeed, if the effect of ticagrelor was mediated by a direct effect at the adenosine receptors, an effect on coronary blood flow with ticagrelor alone would have been expected and this was not observed.
The adenosine-mediated mechanism of action of ticagrelor via inhibition of ENT1 could potentially contribute to the overall clinical benefit of ticagrelor, including the reduction in all cause and cardiovascular mortality observed in ticagrelor-treated patients. 4 For example, adenosine has been shown to have several effects that may be beneficial for patients with ACS, including cardioprotection, vasodilatation, inflammatory regulation, and platelet function inhibition. 8,11 Indeed, local adenosine increases via inhibition of ENT1 could also contribute to the dyspnoea or bradyarrhythmias that have been observed in some patients receiving ticagrelor. 3,4
Of the other P2Y12 antagonists examined, only the cangrelor main metabolite inhibited adenosine uptake at ENT1, with an IC50 of 1.7 µmol/L. No data are available on the plasma concentration of the cangrelor main metabolite following infusion of cangrelor; however it is interesting to note that dyspnoea is also a side effect associated with the use of this agent. 26 –28 No effect was observed for the active metabolite of clopidogrel, consistent with the observation that “adenosine-mediated” effects were not observed in the phase III trials of clopidogrel, 29 although dyspnoea, judged by the investigator to be likely or possibly due to the study drug, occurred in 3.7% of patients receiving clopidogrel and 0% of patients receiving placebo in the ONSET/OFFSET trial. 5 Despite reports of dyspnoea in patients receiving elinogrel at an incidence greater than those receiving clopidogrel, 30 elinogrel demonstrated no significant inhibition of adenosine uptake in transporter-expressing cells in the present study. We were unable to test the demethylated metabolite of elinogrel that has been reported to circulate at a concentration 10% of parent. 31 However, a direct effect for elinogrel on adenosine receptors has not been investigated and can therefore not be excluded. Alternative mechanisms by which all P2Y12 antagonists (including ticagrelor) could induce dyspnoea include an increased release of adenosine triphosphate from red blood cells 32 or nonplatelet P2Y12 receptor-mediated effects (eg, smooth muscle or neuronal P2Y12). 33,34
Conclusion
Ticagrelor inhibits adenosine uptake via inhibition of ENT1. This additional mechanism of action for ticagrelor could potentially be of clinical relevance and help explain the clinical profile of ticagrelor in patients with ACS. Generally, ticagrelor showed low affinity for the adenosine receptor subtypes suggesting that a direct effect of ticagrelor on adenosine receptors at clinically relevant levels is unlikely.
Footnotes
Acknowledgments
The authors would like to thank employees of Ricerca BioSciences LLC, Calliper Life Sciences, and Cerep for performing the receptor binding and functional assays. We thank Drs Maria Belvisi and Mark Birrell of Imperial College, London, for performing isolated vagus experiments. We also thank James Goodman for the generation of the CNT2/3 data and Dr Marie South for statistical advice. Medical writing support was provided by David Evans (Gardiner-Caldwell Communications, Macclesfield, United Kingdom) and this was funded by AstraZeneca. The present studies were funded by AstraZeneca, of which all authors are employees. The data presented in this manuscript have not been published or presented previously.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are employees of AstraZeneca.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: The studies reported in this paper were sponsored by AstraZeneca.