
Abstract
The use of in vitro experimental models of hypoxia-reoxygenation (H/R) that mimic in vivo ischemia-reperfusion represents a powerful tool to investigate cardioprotective strategies against myocardial infarction. Most in vitro studies are performed using neonatal cardiac cells or immortalized embryonic cardiac cell lines which may limit the extrapolation of the results. We developed an H/R model using adult cardiomyocytes freshly isolated from mice and compared its characteristics to the in vivo ischemia-reperfusion conditions. First, cell death was assessed at different values of pH medium during hypoxia (6.2 vs 7.4) to simulate extracellular pH during in vivo ischemia. Cardiomyocyte mortality was aggravated with hypoxia under acidic pH. We next evaluated the relationship between the duration of hypoxia and cell death. Hypoxia time-dependently reduced myocyte viability (−24%, −36%, −53%, and −74% with 1, 1.5, 2, and 3 hours of hypoxia followed by 17 hours of reoxygenation, respectively). We then focused on the duration of reoxygenation as cardioprotective strategies have been reported to have different effects with short and long durations of reperfusion. We observed that cardiomyocyte mortality was increased when the duration of reoxygenation was increased from 2 h to 17 hours. Finally, we used our characterized model to investigate the cardioprotective effect of regular treadmill exercise. Myocyte viability was significantly greater in exercised when compared to sedentary mice (44% and 26%, respectively). Similarly, mice submitted to in vivo ischemia-reperfusion elicited infarct sizes reaching 27%, 43%, and 55% with 20, 30, and 45 minutes of coronary artery occlusion. In addition, infarct size was significantly reduced by exercise. In conclusion, this H/R model of cardiomyocytes freshly isolated from adult mice shows similar characteristics to the in vivo ischemia-reperfusion conditions. The comparison of in vivo and in vitro settings represents a powerful approach to investigate cardioprotective strategies and to distinguish between direct and indirect cardiomyocyte-dependent mechanisms.
Introduction
The development of new cardioprotective strategies remains a major goal to achieve for protecting the heart against myocardial infarction. Currently, particular attention is addressed to the so-called “reperfusion injury.” 1 For this purpose, both in vivo and in vitro experimental models are routinely used. Numerous cardioprotective agents and related cellular mechanisms have been proposed to protect against lethal reperfusion injury. Interestingly, some of them reduce myocardial infarct size in animals during in vivo ischemia-reperfusion while they fail to improve the viability of cardiomyocytes exposed to in vitro hypoxia-reoxygenation (H/R). 2 In this context, the use of isolated cardiomyocytes subjected to hypoxia and reoxygenation may represent a practical tool to distinguish between direct protective effects on the cardiomyocyte and indirect mechanisms, targeting phenomena involving nonmyocytes such as inflammation or vasculoprotection.2,3
The vast majority of in vitro studies have been performed using neonatal cardiac cells and immortalized embryonic cardiac cell lines such as h9c2 cells. Ischemic heart disease, however, is present almost exclusively in the adult population, so the use of adult cardiomyocytes is more physiopathologically relevant. In addition, neonatal cardiomyocytes are more resistant to hypoxia in comparison to adult cells.4,5 Several hypotheses have been raised to explain this phenomenon, for example, differences in ultrastructural profiles, metabolic phenotype, cellular homeostasic regulation (pH-, ion- and redox-regulating systems), or signaling pathways between neonatal and adult cardiac cells.6,7 Importantly, mitochondria, the main organelle involved in cardioprotection, show a potential role in the tolerance of immature heart to oxygen deprivation that could explain the ineffectiveness of cardioprotective strategies when applied to neonatal myocytes. Indeed, it appears that the mitochondrial permeability transition pore (mPTP) of neonates presents a lower sensitivity to pore-opening factors such as calcium (Ca2+). 8 In the adult rat, blockade of mPTP by sanglifehrin A had a protective effect on ischemia-reperfusion injury, 9 whereas it showed no effect in the neonatal heart. 8 Therefore, care must be taken when extrapolating results from embryonic and neonatal myocytes to fully differentiated adult cardiomyocytes.
The present study characterizes a model of H/R using freshly isolated adult mouse cardiomyocytes. The fresh daily isolation overcomes limitations related to cell transformation observed in cultured cells and ensures the preservation of the original phenotype.10,11 Our aims were (1) to define the effects of pH variation during hypoxia on cardiomyocyte mortality as acidosis is one hallmark of the ischemic heart, (2) to examine the effects of increasing durations of hypoxia and reoxygenation on cardiomyocyte mortality, (3) to compare the in vitro characteristics to those of an in vivo model of ischemia-reperfusion in mice, and (4) finally, to validate our in vitro model by investigating the effects of a well-known cardioprotective strategy, that is, regular treadmill exercise.
Methods
Animals
Male 5- to 10-week-old wild-type C57BL/6 J mice (n = 104; R. Janvier, Le Genest St Isle, France) were housed in an air-conditioned room with a 12 hour light–dark cycle and received standard rodent chow and drinking water ad libitum. The authors were granted a license from the institutional office “Préfecture du Val de Marne” (France) to conduct animal research (agreement A94-028-28). The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Isolation of Adult Cardiomyocytes
Animals were anesthetized with an intraperitoneal injection of sodium pentobarbital (90 mg/kg) and heparin (500 IU). The heart was quickly excised, placed into an ice-cold Ca2+-free perfusion buffer (mmol/L: sodium chloride [NaCl] 113, potassium chloride [KCl] 4.7, KH2PO4 0.6, Na2HPO4 0.6, magnesium sulphate [MgSO4] 1.2, NaHCO3 12, KHCO3 10, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES] 10, taurine 30, 2,3-butanedione monoxime 10, glucose 5.5, pH 7.4; Sigma, Saint-Quentin Fallavier, France), and cannulated for retrograde perfusion using a Langendorff apparatus at constant pressure. The heart was first perfused with this perfusion buffer gassed with 95% O2, 5% CO2 for 2 to 3 minutes at 37°C. Then a digestion buffer containing Ca2+ (12.5 µmol/L), liberase thermolysin medium (0.2 mg/mL; Roche Diagnostics, Meylan, France) and trypsin (0.028 mg/mL; Sigma) was used. Once enzyme digestion of the heart was completed, the left ventricle was dissected. It was then minced with scissors and homogenized with a pipette in a stopping buffer containing Ca2+ (12.5 µmol/L) and 10% bovine calf serum (Gibco, Invitrogen Corporation, Cergy-Pontoise, France). The resulting solution was filtered through a gauze and allowed to form a pellet by gravity. The cellular pellet was washed 6 times with the perfusion buffer containing 5% bovine calf serum and increasing concentrations of Ca2+ (12.5, 62, 112, 212, 500, 1000 µmol/L). With this method, we routinely obtained a high percentage (80%) of rod-shaped myocytes with clear striations and without visible vesicles. Only 1 mouse (ie, 1 individual isolation) was used for each experimental condition. Cardiomyocytes obtained from different hearts were never pooled.
Hypoxia-Reoxygenation in Adult Mice Cardiomyocytes
During hypoxia, substrate (glucose/serum) and oxygen deprivation were achieved by series of changes in the medium. Freshly isolated cardiomyocytes were resuspended in serum-free/glucose-free HEPES-buffered medium (mmol/L: NaCl 113, KCl 4.7, HEPES 10, MgSO4 1.2, taurine 30, calcium chloride [CaCl2] 1, pH 7.4 and 37°C). Cells were then allowed to settle for 10 minutes. The supernatant was withdrawn and replaced with the serum-free/glucose-free HEPES-buffered medium bubbled with nitrogen (N2) and adjusted to a chosen pH. Petri dishes (35 mm) were filled with 1 mL of this final cellular suspension and incubated in an O2/CO2 incubator containing a humidified atmosphere of 1% O2, 5% CO2, and 94% N2 at 37°C. After hypoxia, the cells were reoxygenated with 1 mL of HEPES-buffered medium supplemented with bovine calf serum (10%) and glucose (11 mmol/L) bubbled with O2 (pH 7.4) and were placed under normoxia.
In order to provide control conditions, freshly isolated cardiomyocytes were washed twice with a HEPES-buffered medium (mmol/L: NaCl 113; KCl 4.7, HEPES 10, MgSO4 1.2, taurine 30, CaCl2 1, pH 7.4, and 37°C) supplemented with bovine calf serum (5%) and glucose (5.5 mmol/L). Petri dishes were filled with 1 mL of this final cellular suspension and the cells were placed under normoxia. Reoxygenation was mimicked by adding 1 mL of the medium.
Measurement of Cell Viability
At the end of the sequence of H/R or normoxia, cardiomyocyte survival was measured by staining cells with a 0.4% Trypan Blue Solution (Sigma). Cardiomyocytes were visualized under the microscope. Ten fields were analyzed at ×20 magnification and at least 300 cells were counted in each dish. The number of viable (unstained) and nonviable (blue stained) cells was recorded and the percentage of viability was defined as the number of unstained myocytes divided by that of total cells. Survival was expressed as the percentage of the normoxic control values which were set at 100%.
Myocardial Infarction
Mice were anesthetized by intraperitoneal injection of sodium pentobarbital (90 mg/kg), intubated, and ventilated mechanically. The body temperature was maintained at 37°C. A left thoracotomy was performed in order to enable coronary artery occlusion with a 7-0 Prolene thread placed around the left coronary artery and followed by 24 hours of reperfusion as previously described. 12 Myocardial ischemia was confirmed by the occurrence of regional cyanosis. Reperfusion was confirmed by visualization of a hyperemic response and the chest was closed in layers. At the end of the 24-hour reperfusion period, mice were reanesthetized, the coronary artery was reoccluded at the previous site of occlusion, and the heart was excised after Evan blue perfusion. The area at risk was identified using Evan blue staining and the infarct area was identified by 2,3,5-triphenyltetrazolium chloride (TTC) staining. The area at risk was identified as the nonblue region and expressed as a percentage of the left ventricle weight. The infarcted area was identified as the TTC negative zone and expressed as a percentage of area at risk.
Regular Treadmill Exercise
Mice were randomly subjected to regular treadmill exercise or to sedentary conditions. Exercised animals ran on 5 of 7 days during 4 weeks. The first week was an adaptation period with a gradual increase in speeds (10, 14, 18, 22, 26, and 30 cm/s, 4° slope, 5 minutes each step) for 30 minutes per day. Mice ran daily for 1 hour for the following 3 weeks (30 cm/s, 4 slope). No exhaustion was observed and exercise was well tolerated by the mice.
Protocols
Three sets of experiments were used to characterize the in vitro model of H/R (Figure 1). First, protocol A aimed at evaluating the effect of hypoxia on cell survival under acidic conditions versus physiological pH. During hypoxia (1.5 hours), cardiomyocytes were placed at pH 5.4, 6.2, or 7.4 and subsequently reoxygenated at pH 7.4 for 17 hours. Second, protocol B investigated increasing durations of hypoxia performed at pH 6.2 (1, 1.5, 2, and 3 hours) followed by 17 hours of reoxygenation. Third, protocol C studied 2 durations of reoxygenation (2 or 17 hours), hypoxia being performed at pH 6.2 for 3 hours.
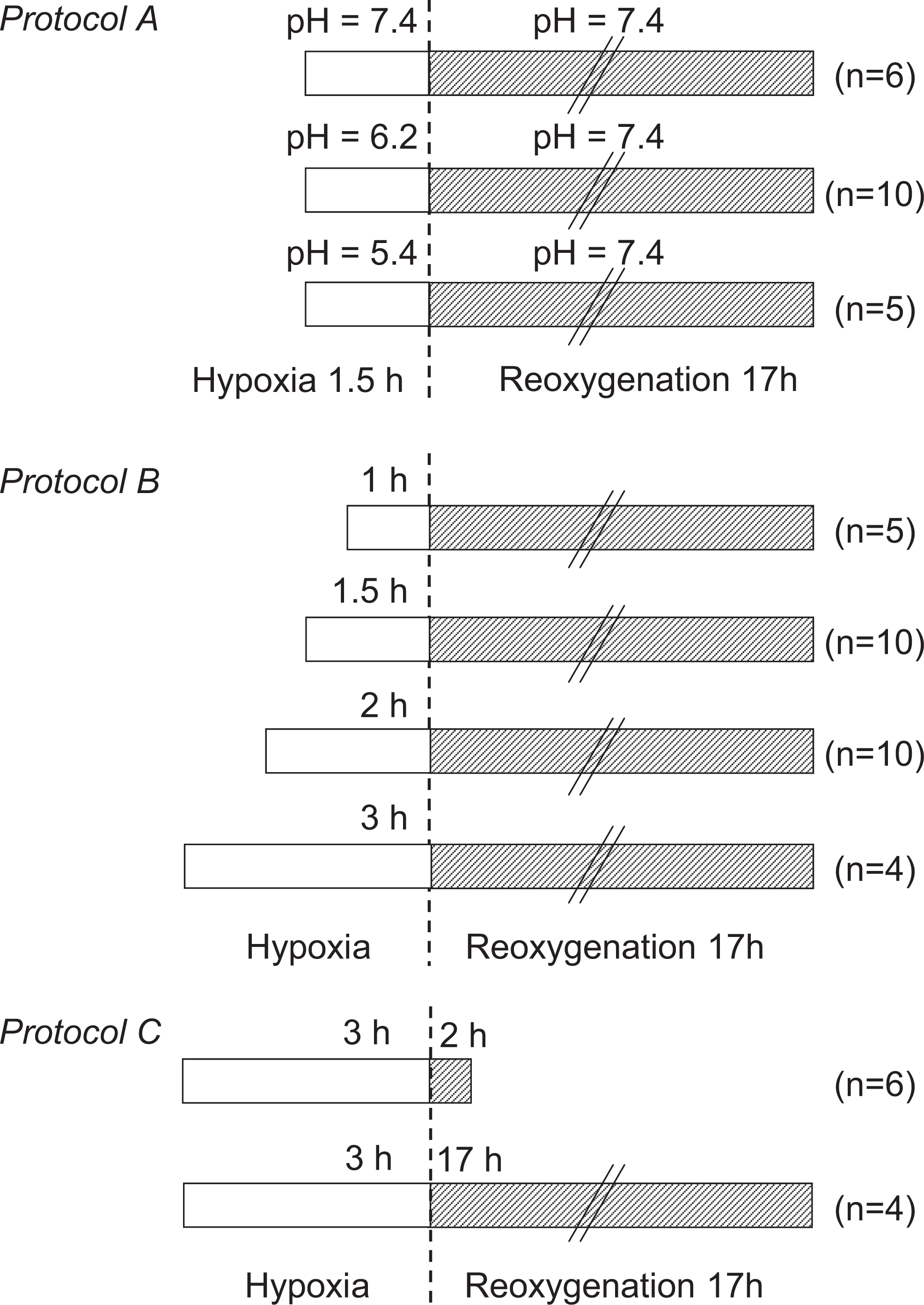
Experimental protocols investigating the effect of (1) pH variation during hypoxia (protocol A), (2) increasing durations of hypoxia (protocol B), and (3) the duration of reoxygenation on cardiomyocyte viability (protocol C). “n” indicates one experiment derived from one heart.
To verify that this model allows detection of cardioprotective activity, we investigated cardiomyocytes collected from mice subjected to regular exercise. Cardiomyocytes were isolated from sedentary and exercised mice. They were exposed to 3 hours of hypoxia at pH 6.2 followed by 17 hours of reoxygenation.
The final objective of this study was to verify that the in vitro and in vivo models elicit similar characteristics. First, as the effects of increasing durations of hypoxia were examined, mice were submitted to different durations of coronary artery occlusion (20, 30, or 45 minutes). Second, exercised and sedentary mice underwent 30 minutes of coronary artery occlusion followed by 24 hours of reperfusion.
Statistical Analysis
All values are expressed as mean ± standard error of the mean. Comparisons were performed using a Kruskal-Wallis analysis followed by Mann-Whitney tests. Statistical significance was defined as a value of P < .05.
Results
Effect of pH Variation During Hypoxia on Adult Cardiomyocyte Viability
Loss of viability of adult cardiomyocytes was assessed at different extracellular pH values during hypoxic incubation (protocol A). The first series of cardiomyocytes was placed in a solution adjusted to pH 7.4 during hypoxia. Using a second set of cells, the pH was adjusted to 6.2 during hypoxia to simulate the acidic environment of the myocardium during ischemia. As illustrated in Figure 2, cardiomyocytes exposed to hypoxia at pH 7.4 (1.5 hours) followed by reoxygenation at pH 7.4 (17 hours) exhibited a 26% ± 3% loss in viability compared to normoxia (absolute cell viability: 68% ± 2%; n = 6). Under acidic conditions, that is, at pH 6.2 during hypoxia, cell death increased to 36 ± 3% of normoxia (absolute cell viability: 68% ± 1%; n = 10). Cell mortality further increased when pH was even lower (pH 5.4) with an 81% ± 3% reduction in viability (n = 5). Thus, in this model of H/R, the return of pH from acidic (hypoxia) to physiologic conditions (reoxygenation) exacerbated lethal injury, in accordance with the well-known phenomenon of the pH paradox. The acidic condition (pH 6.2) during hypoxia was used for protocols B and C.

Effect of pH variation during hypoxia on adult cardiomyocyte viability. Cardiomyocytes were exposed to a sequence of hypoxia at pH 7.4 (n = 6) or 6.2 (n = 10) for 1.5 hours followed by 17 hours of reoxygenation at pH 7.4. Control cells were incubated in complete medium under normoxic conditions for 18.5 hours (normoxia). * P < .05 versus normoxia; † P < .05 versus hypoxia at pH 7.4.
Effect of Increasing Durations of Hypoxia
The next goal was to investigate cell mortality with various durations of hypoxia (protocol B). As illustrated in Figure 3, hypoxia significantly reduced myocyte viability in a time-dependent manner (percent change from absolute cell viability during normoxia: −24% ± 3% from 68% ± 1%, n = 5; −36 ± 3% from 68% ± 1%, n = 10; −53% ± 2% from 70% ± 2%, n = 10 and −74% ± 5% from 70% ± 3%, n = 4 with 1, 1.5, 2, and 3 hours of hypoxia, respectively).
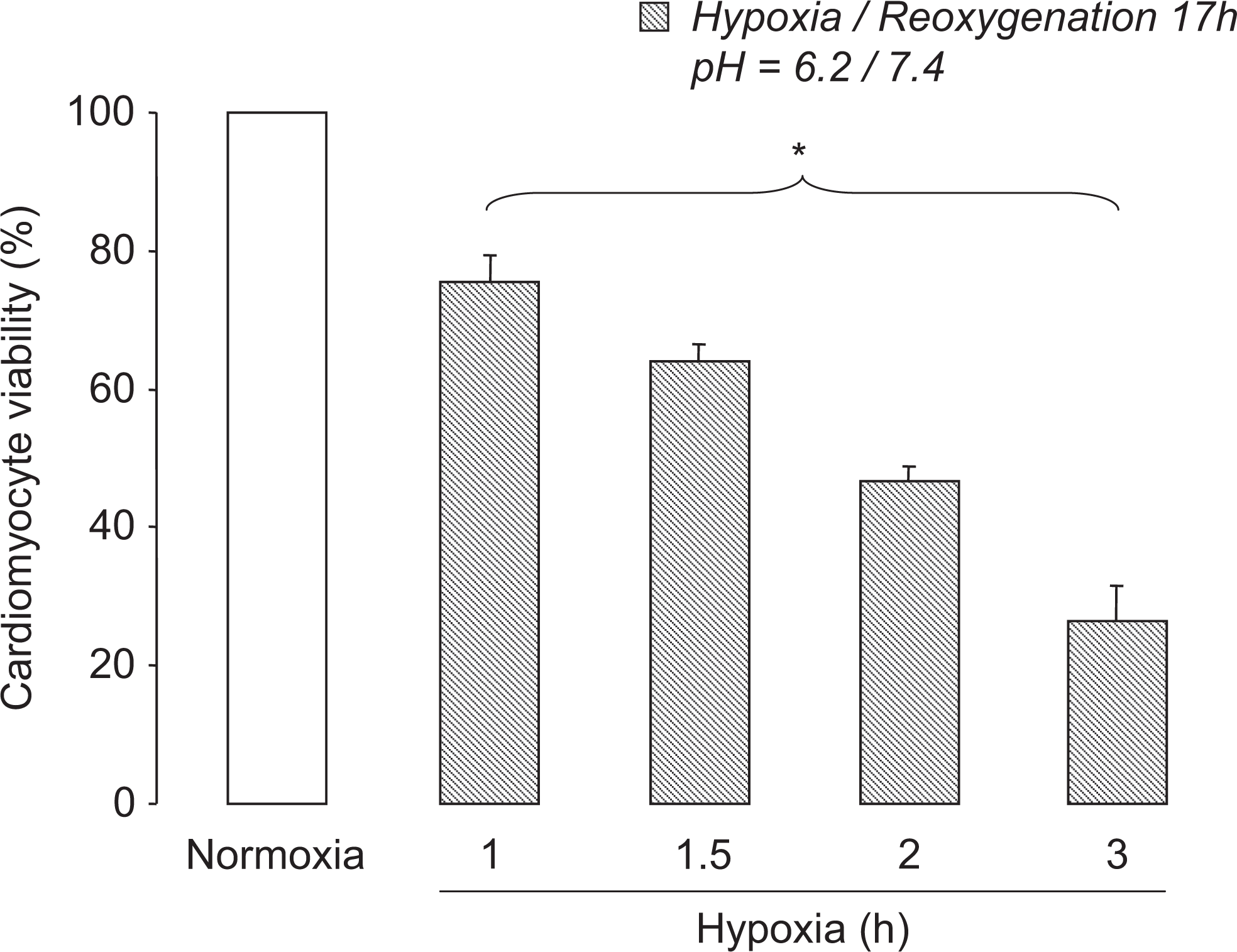
Effect of increasing the duration of hypoxia on cardiomyocyte viability. Cardiomyocytes were exposed to a sequence of hypoxia at pH 6.2 for 1 hour (n = 5), 1.5 hours (n = 10), 2 hours (n = 10), or 3 hours (n = 4). Cells were then reoxygenated and cell viability was determined after 17 hours. Control cells were incubated in complete medium under normoxic conditions for 18 to 20 hours (normoxia). * P < .05 for overall differences.
This relationship between the duration of hypoxia and cell viability observed in vitro mirrored the infarct size response to increasing periods of ischemia in vivo. As expected, prolonging the duration of coronary artery occlusion followed by reperfusion resulted in a significant increase in infarct size that reached 27% ± 4%, 43% ± 3%, and 55% ± 2% with 20, 30, and 45 minutes of coronary artery occlusion (n = 7, n = 7, and n = 8, respectively, Figure 4) with similar areas at risk (42% ± 2%, 43% ± 1%, and 44% ± 2%, respectively). Thus, this model of H/R mimicked the in vivo results.

Relationship between infarct size and increasing the duration of myocardial ischemia. Infarct sizes (expressed as percentage of area at risk) were measured in mice submitted to 20 minutes (n = 7), 30 minutes (n = 7), or 45 minutes (n = 8) of coronary artery occlusion followed by 24 hours of reperfusion. * P < .05 for overall differences. Representative pictures of infarcted myocardium treated with 2,3,5-triphenyltetrazolium chloride (TTC) staining (in white) are shown. The blue area represents the nonischemic zone.
Effects of Increasing the Duration of Reoxygenation
We next focused on reoxygenation and investigated the effects of 2 durations of reoxygenation following 3 hours of hypoxia (protocol C, Figure 5). Cardiomyocyte viability significantly decreased as the duration of reoxygenation was prolonged (percent change from absolute cell viability during normoxia: −31% ± 1%, from 78% ± 2%, n = 6 and −74% ± 5% from 70% ± 3%, n = 4 with 2 and 17 hours of reoxygenation, respectively). This highlights the importance of longer durations of in vitro reoxygenation to reach a more complete extent of injury during H/R.

Effect of increasing the duration of reoxygenation on cell viability. Cardiomyocytes were exposed to a sequence of hypoxia at pH 6.2 for 3 hours followed by 2 hours (n = 6) or 17 hours (n = 4) of reoxygenation. Control cells were incubated in complete medium under normoxic conditions for 5 hours or 20 hours (normoxia). * P < .05 versus normoxia; † P < .05 versus 2 hours of reoxygenation.
Protective Effect of Regular Treadmill Exercise
We finally applied this in vitro model of H/R to investigate the well-known cardioprotective effect of regular treadmill exercise. Cardiomyocytes isolated from hearts of sedentary and exercised mice were exposed to 3 hours of hypoxia (pH 6.2) followed by 17 hours of reoxygenation. In these conditions, the viability of cardiomyocytes was significantly higher in exercised when compared to sedentary mice (absolute cell viability values: 32% ± 5% vs 75% ± 2% for H/R and normoxia, respectively, in exercised mice, n = 4; 18% ± 3% vs 70% ± 3% for H/R and normoxia, respectively, in sedentary mice, n = 4, Figure 6).
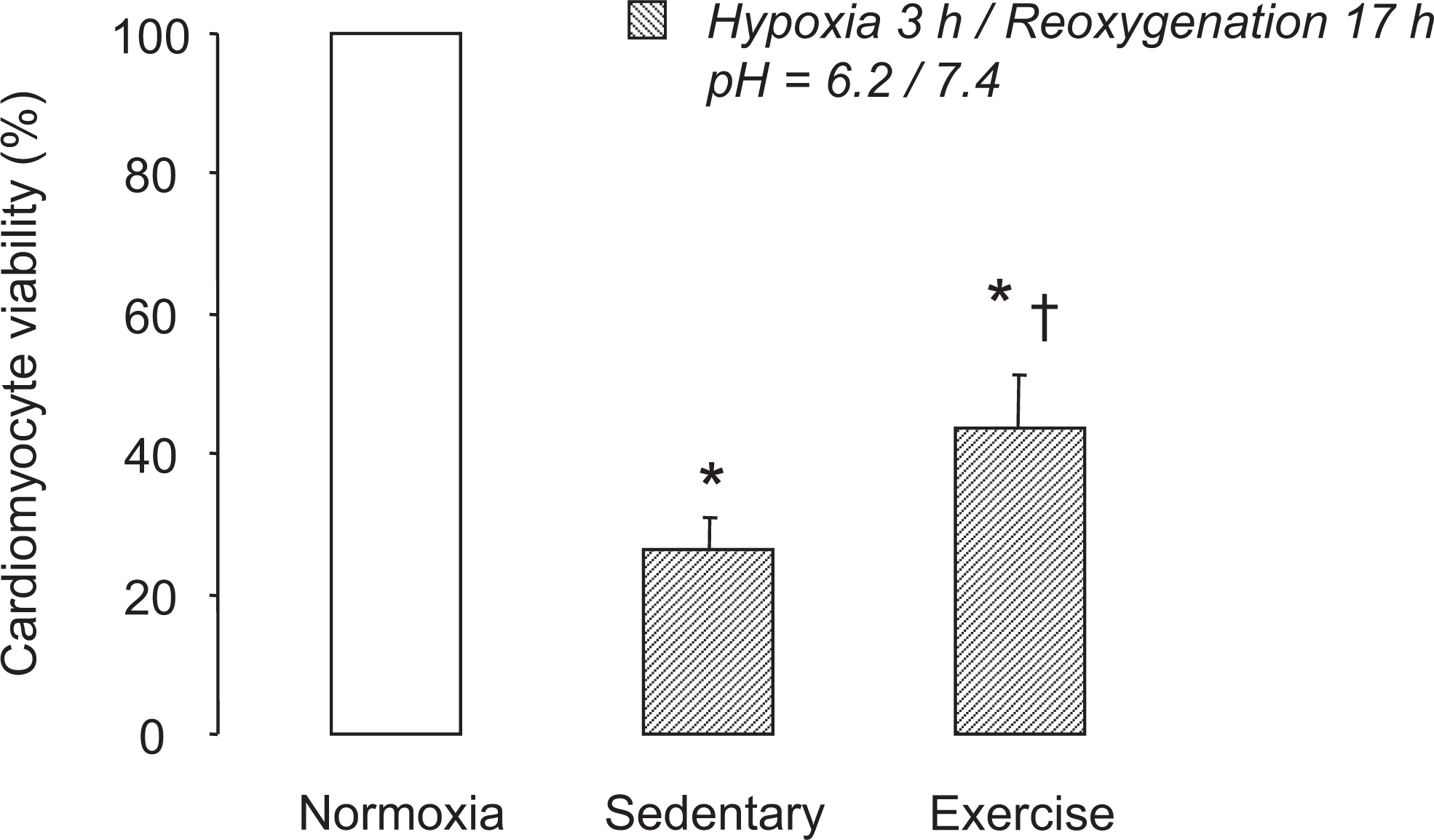
Protective effect of regular treadmill exercise against hypoxia reoxygenation of cardiomyocytes. The viability of cardiomyocytes isolated from heart from sedentary (n = 4) and exercised (n = 4) mice was evaluated after a sequence of 3 hours of hypoxia followed by 17 hours of reoxygenation. * P < .05 versus normoxia; † P < .05 versus sedentary.
In vivo experiments demonstrated a similar pattern in exercised and sedentary mice submitted to 30 minutes of coronary artery occlusion followed by 24 hours of reperfusion. As illustrated in Figure 7, regular exercise (n = 7) allowed a significant reduction of infarct size (−64%) when compared to sedentary animals (n = 7). Taken together, these results verify that this in vitro model of H/R can demonstrate a cardioprotective effect of an appropriate intervention in adult cardiomyocytes.

Cardioprotective effect of regular treadmill exercise against ischemia-reperfusion in mice. Infarct sizes (expressed as percentage of area at risk) were measured in sedentary and exercised mice (both n = 7) submitted to 30 minutes of coronary artery occlusion followed by 24 hours of reperfusion. * P < .05 versus sedentary. Representative pictures of infarcted myocardium treated with 2,3,5-triphenyltetrazolium chloride (TTC) staining (in white) are shown. The blue area represents the nonischemic zone.
Discussion
The present study characterizes an in vitro model of H/R using freshly isolated adult mice cardiomyocytes. An original aspect of the present work was to demonstrate that this model presents similar features to in vivo ischemia-reperfusion model. First, we demonstrated a relationship between the duration of hypoxia and cell viability. This allows the possibility of investigating the ceiling effect of any proposed strategy. Second, we emphasized the importance of the duration of reoxygenation for an extensive evaluation of the cardioprotective potency. Finally, we used our experimental setting to demonstrate the cardioprotected phenotype of cardiomyocytes isolated from mice subjected to regular treadmill exercise.
This in vitro model is characterized by a capacity to control the physical and chemical environment of the cells, making it easier to mimic a wide range of cardiac pathological situations such as ischemia-reperfusion. It should be stressed that multiple models of in vitro cell death have been developed by other groups and the present model is based on these previous findings. In our experiments, hypoxia was accompanied by glucose and serum deprivation to reduce cellular viability during hypoxia in accordance with other groups.13–15 The lack of glucose and fatty acids together with severe hypoxia forces the cardiomyocytes to rely largely on glycogenolysis for adenosine triphosphate (ATP) production which contributes to the cell weakening process. 16 In order to define the number of wash outs sufficient to starve cardiomyocytes at the time of hypoxia, without triggering cardiomyocytes preconditioning, we tested several combinations of maneuvers (data not shown) and decided to perform one wash out to remove substrate and a second to withdraw O2 by adding a medium previously bubbled with N2 just prior to hypoxia. Irreversible metabolic blockade with inhibitors such as cyanide may be inappropriate in simulated ischemia as it complicates the interpretation of cell mortality on simulated reperfusion. At reoxygenation, we supplied the medium with O2 and substrates to simulate the hyperemic response that occurs in vivo at reperfusion.
Severe acidity of the culture medium (pH 6.2) was used to approximate values for extracellular pH during in vivo ischemia in accordance with other reports.15,17 Acidosis is one hallmark of the ischemic myocardium usually attributed to anaerobic glycolysis and release of protons secondary to ATP breakdown. 18 As ischemia progresses, unbuffered intracellular protons can leave ischemic cells through sarcolemmal transporters such as the Na+/H+ exchanger and the H+-lactate cotransporter which induce extracellular acidification. As the extracellular buffering capacity is rapidly overtaken, the proton efflux tends to be impaired thereby reinforcing intracellular acidosis. 19 In our in vitro experimental setting, glucose deficiency and the acidic environment tend to inhibit anaerobic glycolysis, 20 and we can hypothesize that an excess of protons is predominately induced by net ATP breakdown. During hypoxia, we simulated the limited buffering capacity of the interstitium by reinforcing acidity of the extracellular medium (pH 5.4 or 6.2). This imposes an inverse trans-sarcolemmal proton gradient that would alter proton efflux from cardiomyocytes. It has been reported that during hypoxia when cardiomyocytes are surrounded by a buffer solution adjusted to pH 6, the cells exhibit a characteristic drop in intracellular pH (7.1- 6.3) similar to that observed in vivo during ischemia.17,21 In addition, intracellular acidosis during hypoxia promotes apoptosis, whereas minimal DNA fragmentation is observed when acidosis is corrected with sodium hydroxide or HEPES. 22 Conversely, intracellular acidification of cardiac myocytes through inhibition of vacuolar proton ATPase by bafilomycin A1 is accompanied by apoptotic cell death. 23 Moreover, some cardioprotective strategies, such as preconditioning, reduce intracellular acidification during ischemia notably by activating vacuolar proton ATPase and thereby extruding protons. 24
To simulate reperfusion, we reoxygenated cells under physiological pH. In accordance with others groups,17,25 we found that incubating cardiomyocytes under acidic pH during hypoxia exacerbates cell mortality at reoxygenation. Indeed, the return of intracellular pH from acidic to physiologic values at reoxygenation precipitated cell killing defining the phenomenon of “pH paradox.” The latter represents an independent event aggravating injury and causing cell death since reoxygenation at acidic pH prevents cardiomyocyte damage.26–28 One of the hypotheses to explain this paradox is that normalization of intracellular pH could act as a permissive factor for hypercontracture. Indeed, intracellular pH recovery contributes to Ca2+ overload by concomitant activation of Na+/H+ and Na+/Ca+ exchangers. 29 Acidosis is also known to prevent mPTP formation by blocking Ca2+ binding to adenine nucleotide translocase and displacing cyclophilin D from it.30,31
In our experimental conditions, we found that the severity of the cell death was influenced by the duration of hypoxic insult. The longer the duration of hypoxia, the greater the magnitude of cell death. This mimics in vivo ischemia-reperfusion conditions as it is well known that prolongation of ischemia is responsible for a time-dependent increase in infarct size. 32 The possibility of investigating several durations of ischemia facilitates exploration of the ceiling effect of a cardioprotective strategy.33–35 In our in vitro conditions, this can be translated as the duration of hypoxia and the extent of cell damage at which beneficial effects are lost. It is also important to emphasize the critical role of reoxygenation duration on cell injury. We observed that cardiomyocyte mortality was aggravated as the duration of reoxygenation was increased in accordance with previous results. 36 It might be relevant to test the cardioprotective effect of drugs with a prolonged duration of reoxygenation in order to avoid false positive results. Previous in vivo reports have clearly demonstrated that the beneficial effects of several cardioprotective strategies were observed with short but not with long durations of reperfusion.37,38 In contrast, it is still possible to reduce myocardial infarct size when a drug or strategy is applied late during reperfusion39–42 suggesting that long periods of reperfusion are needed to observe the final extent of damage and to determine the real potency of a cardioprotective strategy.
We used Trypan blue to assess cell viability. Blue cell staining indicates breakdown of the plasma membrane permeability barrier. This classical method of cell death identification continues to be widely employed and is advantageous in that it limits false estimation by the examination of staining and structural alterations, dead cells being round shaped. Other methods are based on the precise quantification of a single cellular process and may be excessively specific and thus result in the over and/or underestimation of cell mortality. For instance, the tetrazolium salt assay reflects the activity of the mitochondrial chain. Quiescent and viable cells, however, are not always distinguished from dead cells, thus underestimating cell viability. 43 Further, the reduction of tetrazolium salt to formazan is predominantly mediated by mitochondrial dehydrogenase, but this conversion could be promoted by other cell reduction systems and notably by reactive oxygen species such as superoxide. 43 This may lead to false positive results concerning the estimation of cell viability during H/R. The pharmacological agents tested might also affect the tetrazolium salt reduction per se. 44 Finally, enzyme release techniques such as the lactate dehydrogenase release assay also have several drawbacks. The enzymatic activity tends to decrease with time as a result of natural degradation and can be affected by several variables including medium composition. Although tedious, the direct verification of cardiomyocyte mortality by Trypan blue remains appealing. It should be acknowledged that despite their limitations, additional independent cell viability assays would help to confirm the results.
Finally, using a sequence of 3 hours of hypoxia followed by a prolonged duration of reoxygenation (17 hours), we investigated the capacity of our in vitro H/R model to demonstrate the cardioprotective properties of regular treadmill exercise. Exercise is a well-known, beneficial strategy that protects the heart against ischemia-reperfusion injury through multiple mechanisms involving, among others, vascular and cardiomyocyte adaptations.45,46 Our in vitro setting also highlighted the cytoprotection that mirrored the in vivo reduction in infarct size. We did not specifically investigate the mechanisms of the protective effects of exercise, but one could hypothesize that the cardiomyocyte activation of PI3K-Akt and Erk 1/2 pathways might participate in the cardioprotection afforded by chronic exercise when compared to preconditioning.47,48 These signaling cascades target the mitochondria by inhibiting the opening of the mPTP which leads to cell death. It should be acknowledged that this experimental setting does not consider other possible noncardiomyocyte protective mechanisms such as vasculoprotective effect, and only a single duration of ischemia or hypoxia was investigated. More detailed investigations are needed to determine the cardioprotective mechanism of regular exercise.
In conclusion, this H/R model of cardiomyocytes freshly isolated from adult mice shows similar features to in vivo ischemia-reperfusion conditions. It represents a powerful approach to decipher the molecular and cellular basis for protecting the myocardium and to develop strategies against ischemic injury. The use of isolated cardiomyocytes submitted to hypoxia and reoxygenation associated with the in vivo ischemia-reperfusion model represents a tool to distinguish between direct cardiomyocyte-dependent mechanisms and other indirect effects of cardioprotective strategies.
Footnotes
Acknowledgments
The authors are greatly indebted to Dr Owen Woodman for correcting the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Lolita Portal was a recipient of a grant from the Region Ile de France (CODDIM, 2010) .