
Abstract
Pseudorabies (Aujeszky disease) virus (PRV) was eliminated from domestic swine in many countries using glycoprotein E (gE)-deleted vaccines and serum antibody gE ELISAs, but PRV continues to circulate in some regions and in most feral swine populations in the world. We created a dual-matrix (serum and oral fluid) indirect IgG gE ELISA (iELISA) and evaluated its performance using samples from 4 groups of 10 pigs each: negative control (NC), vaccination (MLV), PRV inoculation (PRV), and vaccination followed by challenge (MLV-PRV). All serum and oral fluid samples collected before PRV challenge and all NC samples throughout the study were negative for gE antibodies by commercial blocking ELISA (bELISA) and our iELISA. Nasal swab samples from 9 of 10 animals in the PRV group were gB quantitative PRC (qPCR) positive at 2 days post-inoculation (dpi). The oral fluid iELISA detected a significant S/P response in the PRV (p = 0.03) and MLV-PRV (p = 0.01) groups by 6 dpi. ROC analyses of serum bELISA (n = 428), serum iELISA (n = 426), and oral fluid iELISA (n = 247) showed no significant differences in performance (p > 0.05). Our data support the concept of PRV surveillance based on oral fluid samples tested by an indirect gE ELISA.
Pseudorabies virus (PRV; Herpesviridae, Varicellovirus, Suid alphaherpesvirus 1) is an enveloped DNA virus. 21 In swine, the clinical expression of PRV infection ranges from acute mortality in piglets to clinically inapparent infection in older animals.33,38,39,44 PRV has been recognized since the 19th century, but an increasing frequency of clinical outbreaks of PRV in commercial swine herds in North America and Europe beginning in the 1960s brought a research focus to the problem of PRV control and eventually led to the development of PRV marker vaccines and differential serum antibody ELISAs.3,16
The development of PRV vaccines began with the recognition that certain viral proteins associated with virulence were not essential for viral replication. This led to the identification of both naturally attenuated (e.g., Bartha K-61, Norden [BUK], and NIA-4) and genetically engineered vaccine strains.11,23 Among vaccine candidates, PRV strains with deletions in the region of the genome encoding for glycoprotein E (gE) were capable of replication, but were less virulent both in cell culture and in pigs. 18 Furthermore, pigs inoculated with gE-deleted viruses developed protective immunity against field viruses and antibody against a range of viral proteins, but did not produce antibody against gE. Thus, it was possible to differentiate infected from vaccinated animals (DIVA) on the basis of the presence or absence of specific antibody. 43 Subsequent research led to the current PRV DIVA approach based on the absence or presence of antibodies against PRV glycoproteins B (gB) and E (gE); gB is essential to virus replication and is induced by either vaccination or infection with wild-type PRV; gE is absent in gE DIVA vaccines but present in wild-type PRVs. Thus, vaccinated and/or uninfected animals (gB+/gE−) can be differentiated from infected animals (gB+/gE+) in an eradication program. These tools formed the foundation of successful campaigns for the eradication of PRV from domestic swine in many countries (e.g., France [2008], Great Britain [1991], Germany [2003], New Zealand [1997], United States [2004]).2,9,13,27,30
PRV continues to present a risk to swine producers because it remains endemic in feral swine populations in Europe, Asia, and the Western Hemisphere.15,19,25,28,29,32,40,41 In the United States, PRV has occasionally been introduced into “transitional” herds via contact with feral swine (e.g., Minnesota [2002], Wisconsin [2007]). In France, PRV was detected in 2019 in swine on 2 farms reportedly via contact with feral swine. 12 Likewise of concern, a PRV variant producing severe clinical signs and high mortality was reported in 2011 in major swine-producing regions of China.1,42,48,49 Isolation of the variant from vaccinated farms raises concerns about the efficacy of contemporary commercial vaccines.1,10 Given these circumstances, improvements in PRV testing, surveillance, control, and elimination remain relevant to global swine health.
Assays based on the detection of nucleic acid and/or pathogen-specific antibodies in swine oral fluid specimens were first described in 200834,35 and, thereafter, used increasingly for disease monitoring and surveillance. 5 PRV antibodies have been detected in swine oral fluids by adapting commercial serum ELISAs (i.e., a PRV gB blocking ELISA [bELISA] and a whole-virus indirect ELISA [iELISA]) to the oral fluid matrix.7,31 Our objective was to further explore the oral fluid–based DIVA approach by developing and evaluating the performance of a PRV oral fluid gE iELISA prototype using swine oral fluid specimens of known vaccination and/or infection status.
Materials and methods
Experimental design
Our prototype gE oral fluid iELISA was created using recombinant antigens generated with a Pichia pastoris (gE) expression system and evaluated using oral fluid and serum specimens from pigs (n = 40) of known PRV vaccination and/or inoculation status. For purposes of comparison, serum specimens were tested for gB and gE antibody using commercial bELISAs (Pseudorabies virus gB antibody test kit, Pseudorabies virus gpI antibody test kit; Idexx). Assay performance was evaluated by receiver operating characteristic (ROC) analysis. All procedures were approved by the Institutional Committees at the USDA National Animal Disease Center (protocol ARS-2017-689) and Iowa State University (protocol 10-17-8622-S).
Pseudorabies virus
The virus that we used (PRV 3CR Ossabaw strain) was propagated and titrated on swine testicular (ST) cells, as described previously.7,50 In brief, confluent monolayers of ST cells in 75-cm2 flasks were inoculated with virus (0.5 mL, 1 × 106.8 TCID50/mL). When cytopathic effect (CPE) was observed in 80–90% of the monolayer, plates underwent 2 freeze–thaw cycles (−80°C, 22°C) to lyse cells and release virus. Thereafter, the solution was centrifuged (1,000 × g; 10 min) and the supernatant recovered.
The virus concentration in the inoculum was determined before and after animal inoculations using standard methods.7,36 In brief, confluent monolayers of ST cells were inoculated with 10-fold serial dilutions of the inoculum, after which plates were incubated for 72 h and inspected for CPE. The TCID50/mL and 95% confidence interval (95% CI) were calculated using the Spearman–Kärber method. 36
Animals, treatments, and sampling
Forty 12–16-wk-old, crossbred pigs were obtained from a porcine reproductive and respiratory syndrome virus (PRRSV; Arteriviridae, Betaarterivirus, Betaarterivirus suid 1 and 2)- and porcine epidemic diarrhea virus (Coronaviridae, Alphacoronavirus, Porcine epidemic diarrhea virus)-free commercial herd in the midwestern United States. After arrival, all pigs were screened for serum antibodies against PRRSV, PRV, and swine influenza A virus using routine methods (Iowa State University, Veterinary Diagnostic Laboratory, Ames, IA, USA). Animals were randomly allocated into 4 groups of 10 pigs: negative control (NC), modified-live vaccination only (MLV), inoculation only (PRV), and vaccination followed by challenge at 21 d post-vaccination (MLV-PRV). The NC group was housed in a biosafety level 2 (BSL-2) facility at Iowa State University; the MLV, PRV, and MLV-PRV groups were housed in BSL-3 facilities under the supervision of the Agricultural Research Services (ARS) at the USDA National Animal Disease Center (Ames, IA, USA). All animals were individually penned, fed, and managed on a daily basis throughout the study. 7
In brief, pigs in the NC group received no treatment and were followed from 0 d of study (DOS) through DOS 49 (Fig. 1). Animals in the MLV and MLV-PRV groups received one dose of a commercial gE-deleted modified-live PRV vaccine (Ingelvac Aujeszky MLV; Boehringer Ingelheim) on DOS 7. On DOS 28, pigs in the PRV and MLV-PRV groups were administered 1 mL of an inoculum containing 1×102.9 TCID50/mL of PRV 3CR Ossabaw into each naris (i.e., 2 mL/pig). Because of BSL-3 space and personnel restrictions, the MLV group was followed through DOS 27 and then replaced by the PRV group through DOS 63.
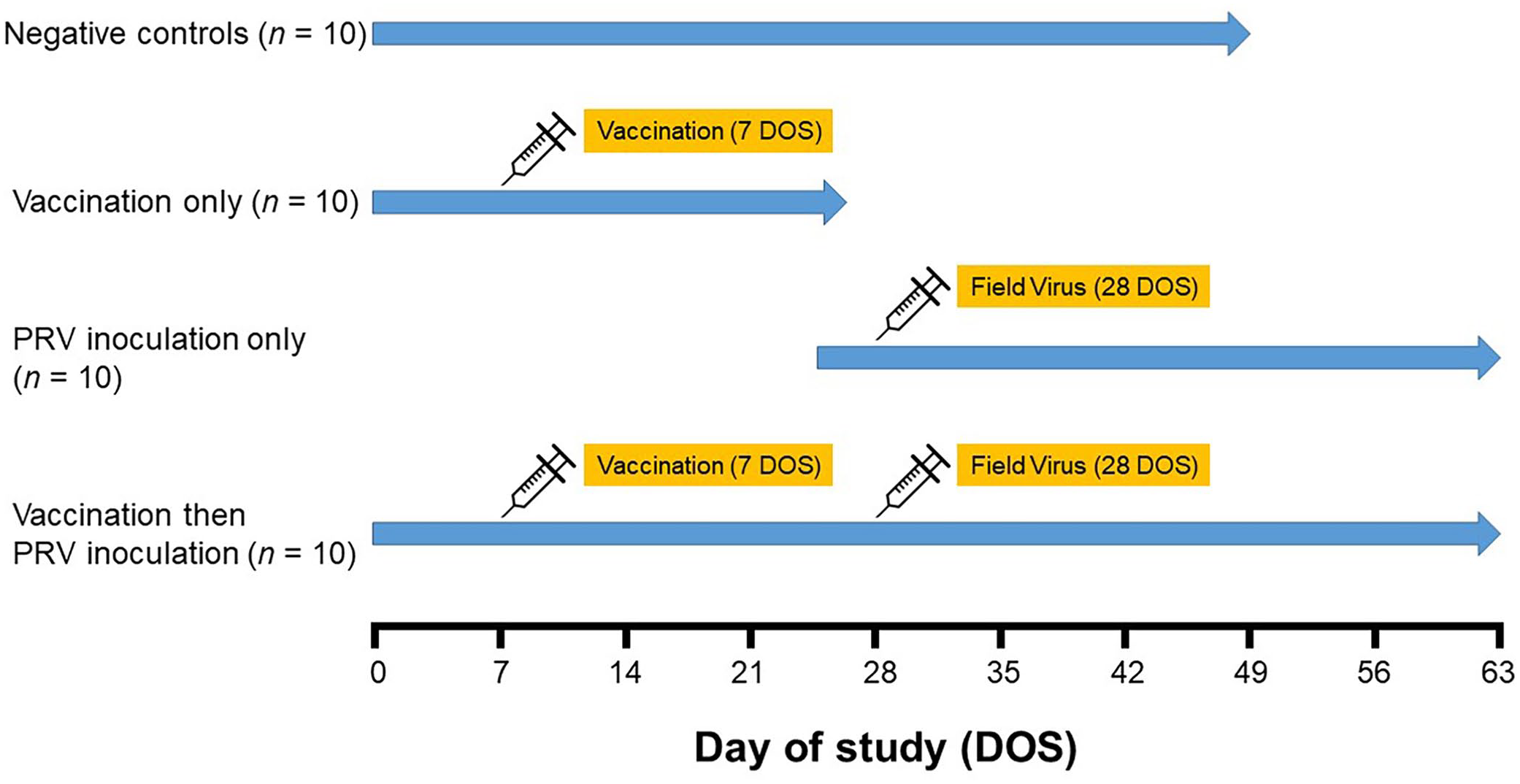
Experimental design for the evaluation of pseudorabies virus (PRV) antibody ontogeny in serum and oral fluid specimens. Negative control group: pigs received no treatment and euthanized on day of study (DOS) 49. Vaccination-only group: pigs vaccinated intramuscularly with a modified-live virus vaccine (Ingelvac Aujeszky MLV; Boehringer Ingelheim) on DOS 7 and euthanized on DOS 27. PRV inoculation–only group: pigs inoculated intranasally with PRV isolate 3CR Ossabaw (1 × 103.5 TCID50/pig) on DOS 28. Vaccination then PRV inoculation group: pigs vaccinated on DOS 7 and challenged on DOS 28.
Blood samples were collected weekly from each animal using disposable 20-ga needles (Exelint), reusable hubs (Becton Dickinson), and 12.5-mL serum separator tubes (Covidien). Thereafter, blood samples were centrifuged (1,000 × g, 15 min), and the separated serum was harvested and stored at –20°C. Nasal swab samples were collected 2, 6, and 9 d after inoculation (DOS 30, 34, 37) from animals in the NC, PRV, and MLV-PRV groups. Each naris was sampled, after which the swab (Puritan Medical Products) was placed in a tube containing 1 mL of phosphate-buffered saline (PBS, pH 7.2; Life Technologies) and stored at −80°C. Oral fluid samples were collected twice daily (morning and afternoon) using cotton rope (Web Rigging Supply) suspended in the pen, as described elsewhere.7,14 In brief, each animal was allowed to interact with the rope for 30 to 45 min. After placing the wet portion of the rope inside a plastic bag, the oral fluid was recovered by passing the rope and bag through a chamois wringer (Dyna-Jet Products). The oral fluid that pooled in the corner of the bag was decanted into 50-mL conical tubes (Falcon) for storage at −20°C.
Each animal was observed twice daily, and a record was maintained of general health and/or clinical signs. At the termination of the study, euthanasia was performed by trained personnel. Animals in the NC group were euthanized by captive bolt; chemical injection was used for the MLV, PRV, and MLV-PRV groups. 17
GE antigen generation and ELISA plate preparation
A codon-optimized gene fragment was selected from the immunodominant regions of the PRV gE protein (Table 1; Fig. 2) for constitutive expression using a P. pastoris expression system and subsequent purification by affinity chromatography, as described previously. 47 The truncated gE gene fragment (codons 31–270) was synthesized with an amino-terminal polyhistidine (His6) tag and amplified by PCR (Table 1). Thereafter, the amplicon was ligated into the P. pastoris expression vector pPICZαA (Invitrogen). The recombinant pPICZαA-gE plasmid vector was linearized with SacI and electroporated (Bio-Rad) into competent P. pastoris X33 strain cells (Thermo Fisher). Thereafter, pPICZαA-gE–positive transformants were cultured for 2–3 d at 30°C on yeast extract–peptone–dextrose (YPD) agar plates (1% yeast extract, 2% peptone, 2% agarose, 2% glucose) containing 800 μg/mL of Zeocin (Thermo Fisher). Colonies resistant to Zeocin were selected and further confirmed to be gE transformants by genomic PCR using a 5′ alcohol oxidase (AOX) primer (5′-GACTGGTTCCAATTGACAAGC-3′), a 3′ AOX primer (5′-GCAAATGGCATTCTGACATCC-3′), and restriction analysis.
DNA and amino acid sequences of the codon-optimized pseudorabies virus gE fragment for protein recombination.

Amino-terminal polyhistidine (His6) tag.
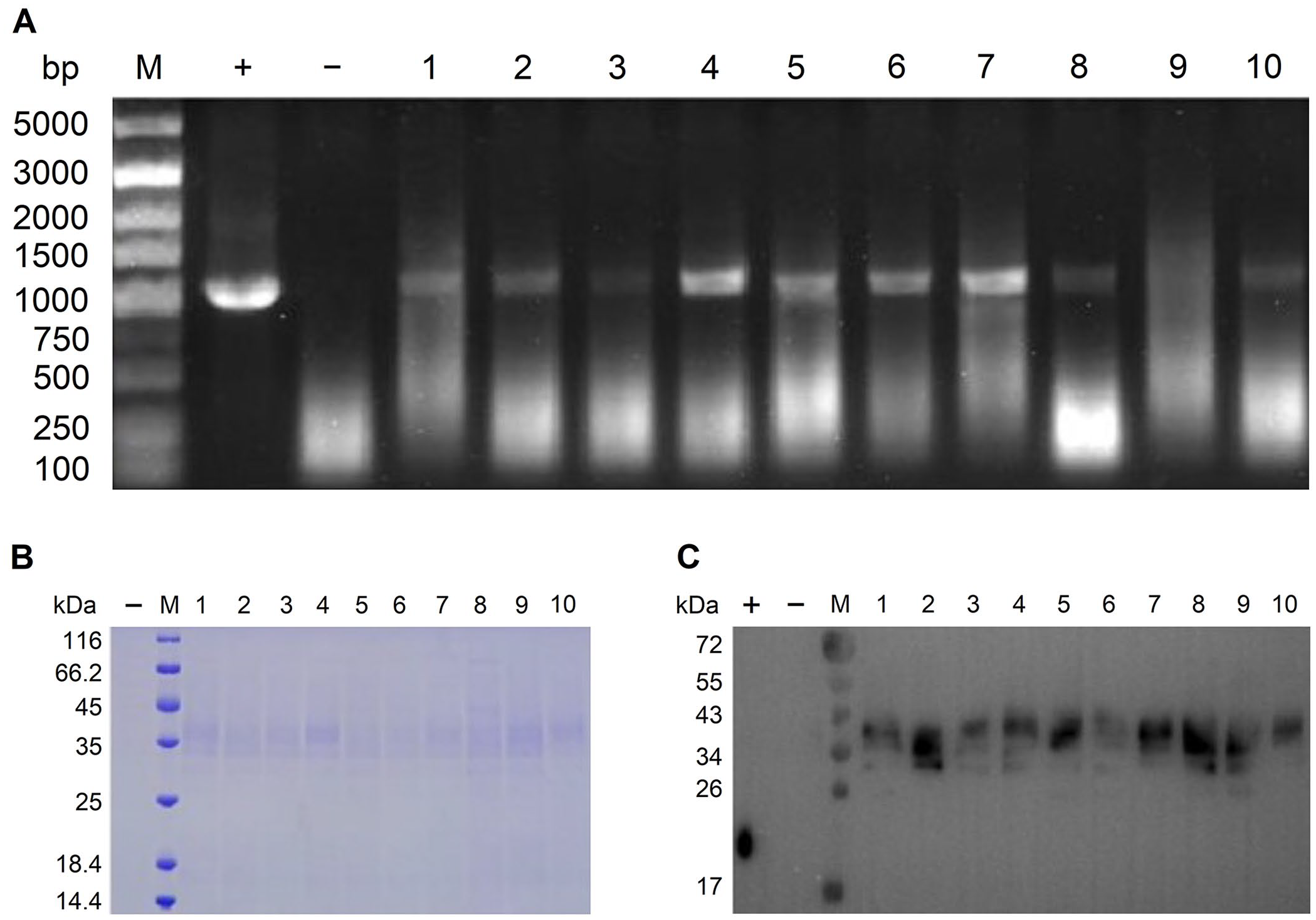
Selection of Pichia pastoris transformed colonies containing the pPICZαA-gE plasmid vector by PCR and gel electrophoresis.
PCR-verified transformants were inoculated into YPD medium containing 1 M sorbitol and shaken vigorously (300 rpm) at 30°C until the culture reached log-phase. Cultured cells were harvested by centrifugation (3,000 × g, 5 min) and resuspended to an optical density of 600 nm (OD600) of ~1 in buffered methanol-complex medium to induce the expression and secretion of target gE protein. Harvested cells were incubated at 30°C; 100% methanol was added to reach a concentration of 1% every 24 h to sustain the induction. After 72 h of induction, the cell culture was harvested by centrifugation and concentrated (10×) for protein expression analysis using sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) and western blot (Fig. 2).
Truncated recombinant gE protein (249 aa; 26.7 kDa) was purified from selected culture using nickel–nitrilotriacetic acid (Ni-NTA) affinity columns (GE Healthcare) according to the manufacturer’s instructions. After thorough washing with wash buffer (PBS, 20 mM imidazole, pH 7.4) the bound gE protein was eluted with elution buffer (PBS, 500 mM imidazole, pH7.4). Protein purification was verified by testing the supernatant, flow through, wash buffer, and eluted sample using SDS-PAGE (Fig. 3). The final protein was dialyzed against PBS (pH 7.4) at 4°C overnight, during which time the dialysis buffer was changed 3 times. The dialyzed recombinant gE protein was analyzed using SDS-PAGE under reducing and non-reducing conditions (Fig. 3). Protein concentration (2.34 mg/mL) was determined using a bicinchoninic acid protein assay kit with bovine serum albumin as a standard (Pierce Biotechnology).
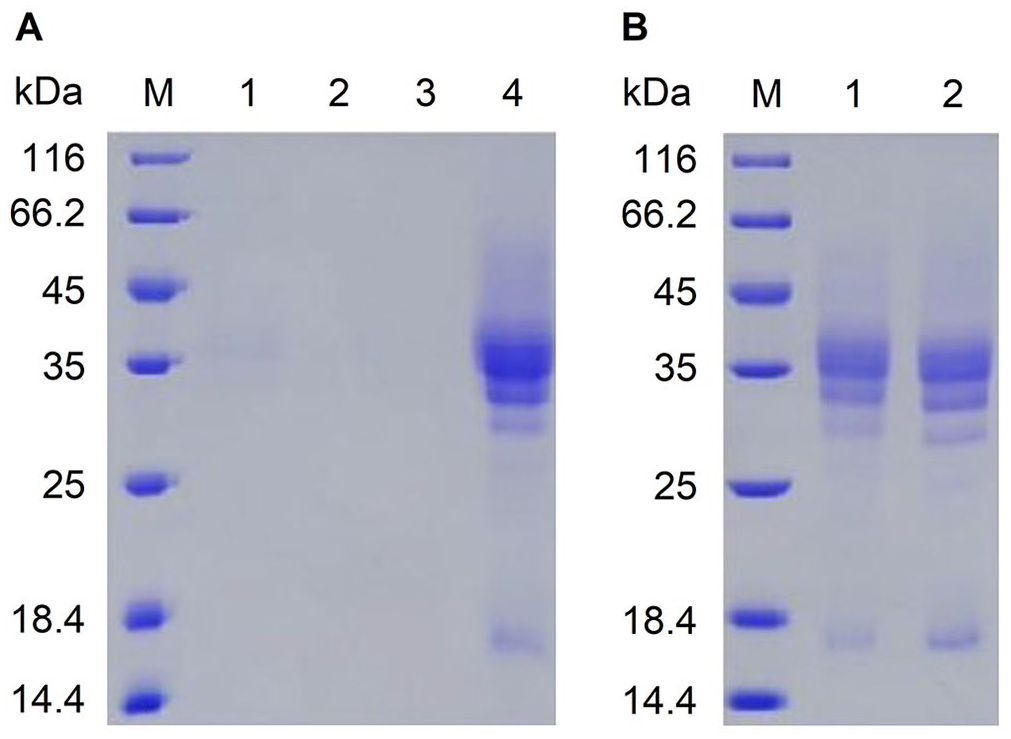
Purification and characterization of the recombinant gE expressed and secreted by Pichia pastoris transformants.
ELISA testing (indirect and commercial)
PRV gE ELISAs for serum and oral fluids were produced by loading 96-well polystyrene plates (Maxisorp; Nunc) with 100 µL per well of PBS (pH 7.4; Gibco, Thermo Fisher) containing recombinant gE (~1.2 µg/mL) and then incubating plates at 4°C for 16 h. The plates were then washed 5 times with PBS containing 0.1% Tween 20 (PBS-T; MilliporeSigma), blocked (150 µL/well) with 1% (w/v) blocking solution (Candor Bioscience), incubated at 20–22°C for 2 h, and dried at 37°C for 4 h.
To perform the IgG gE ELISA, samples (100 µL/well) were diluted 1:100 (serum) or 1:1 (oral fluids) in 50% goat serum–based diluent, incubated at 37°C for 1 h (serum samples) or 2 h (oral fluid samples), and washed 5 times with PBS-T. Thereafter, 100 μL of horseradish peroxidase (HRP)-conjugated goat anti-pig IgG (fragment crystallizable [Fc] region) antibody (1:30,000 for serum samples, 1:1,500 for oral fluid samples; Bethyl Laboratories) was added to each well for serum and oral fluid samples, respectively. Plates were then incubated at 37°C for 1 h and washed 5 times with PBS-T. The antibody–antigen reaction was visualized by adding 100 μL of tetramethylbenzidine–hydrogen peroxide (TMB; Surmodics) substrate solution to each well and incubating for 5 min at 20 to 22°C. The reaction was terminated by adding 100 μL of stop solution (Surmodics) to each well, after which the optical absorbance was measured at 450 nm using an ELISA reader (Molecular Devices). The gE IgG antibody responses were expressed as sample-to-positive (S/P) ratios (equation 1).

Serum samples were also tested using a commercial gE blocking ELISA (Pseudorabies virus gpI antibody test kit; Idexx). The testing procedures provided by the manufacturer were followed, and the results were expressed as sample-to-negative (S/N) ratios (equation 2). Samples with S/N ratios ≤0.6 were classified as positive and >0.7 as negative.

PRV gB qPCR
Nasal swab samples from NC, PRV, and MLV-PRV groups were tested using a PRV gB quantitative PCR (qPCR) to verify the pigs’ infection status using established protocols.8,20 In brief, genomic DNA was extracted from nasal swab samples (MagMax pathogen RNA/DNA kit; Life Technologies) on the KingFisher flex purification system (Thermo Fisher) using modified lysis procedures. 4 Each sample, including nuclease-free water blanks (reference amplification controls), was spiked with 0.5 µL of a synthetic nucleic acid internal positive control (IPC) during the extraction procedure to monitor extraction and qPCR procedures. Each PRV gB qPCR reaction contained 5 µL of TaqMan fast virus 1-step master mix (Thermo Fisher), 7.45 µL of nuclease-free water (Invitrogen), 5 µL of DNA extract, 0.8 µL of 10 µM PRV gB forward primer (5′-ACAAGTTCAAGGCCCACATCTAC-3′), 0.8 µL of 10 µM PRV gB reverse primer (5′-GTCYGTGAAGCGGTTCGTGAT-3′), 0.4 µL of 10 µM PRV gB probe (FAM-5′-ACGTCATCGTCACGACC-3′-BHQ1), 0.2 µL of 10 µM IPC forward primer (5′-TTCGGCGTGTTATGCTAACTTC-3′), 0.2 µL of 10 µM IPC reverse primer (5′-GGGCTCCCGCTTGACAATA-3′), and 0.15 µL of 10 µM IPC probe (Cy5-5′-CTCCGCAGA-TAO-AATCCAGGGTCATCG-3′-IAbRQSp). Extracted PRV Shope strain DNA was included in each qPCR run as a positive amplification control (PAC). In addition, purified PRV gB nucleic acid and nuclease-free water were included as positive and negative amplification controls in each qPCR reaction. Samples with IPC cycle quantification (Cq) values <40 were considered valid. Samples with qPCR gB Cq values <40 were interpreted as positive for PRV.
Statistical analysis
For the analysis of gE iELISA antibody, serum and oral fluid S/P responses in PRV-treated animals (MLV, PRV, and MLV-PRV) were compared to the NC group by DOS using the Wilcoxon rank sum test. Thereafter, the diagnostic sensitivities and specificities of the serum and oral fluid iELISAs were derived from ROC curves calculated using the Comprehensive R Archive Network (CRAN) package pROC (v.1.16.2; https://cran.r-project.org/web/packages/pROC/index.html). 37 For the ROC analysis, the “true” classification of each data point was established using prior PRV studies.7,31 Because of the use of a gE-deleted vaccine, serum and oral fluid samples collected ≤7 d post-inoculation (dpi) were classified as PRV gE antibody negative and those collected ≥14 dpi were considered gE antibody positive. Confidence intervals (95% CI) for diagnostic sensitivities and specificities were estimated as described previously for correlated data. 7 In brief, 1/7 power transformed S/P values were fitted into a multivariate linear mixed model for the estimation of variances while accounting for data correlation.
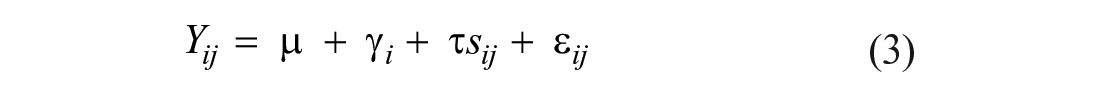
In equation 3, Yij is the jth observation for the ith subject; µ is the overall mean for samples classified as PRV antibody negative; γ i is the random effect of the ith subject; τ is the fixed effect indicating the mean difference between PRV antibody-negative and -positive groups; sij is the disease status of the jth observation for the ith subject; and ε ij is the random error of the jth observation for the ith subject. The 95% CIs for diagnostic sensitivity and specificity based on specific cutoffs were estimated using the variances estimated from equation 2. Logit-transformation was implemented to maintain the estimates within the boundaries of 0 and 1.
Area under curve (AUC) 95% CIs and AUC pairwise comparisons (serum bELISA vs. serum iELISA vs. oral fluid iELISA) were calculated using a nonparametric stratified bootstrap method with 10,000 iterations. 24
Results
Nasal swab samples from 9 of 10 animals in the PRV group were gB qPCR positive with (Cq) values of 30.4–38.2 at 48 h post-inoculation. The last detection of PRV gB DNA was on 35 dpi. One animal in the MLV-PRV group was qPCR positive at 48 h post-inoculation, and all samples from the NC group were qPCR negative. Clinical signs consistent with PRV infection (e.g., anorexia, ataxia, tremors, were observed in 3 of 10 animals in the PRV group at 7 dpi), with all animals clinically normal by 14 dpi.
All serum and oral fluid samples collected before PRV challenge and all NC samples throughout the study were negative for gE antibodies by both bELISA and iELISA. In the PRV group, 3 of 10 animals were positive on the serum bELISA at 13 dpi (Fig. 4). In contrast, 1 of 10 animals in the MLV-PRV group was serum bELISA positive on day 20 post-challenge (Fig. 4).
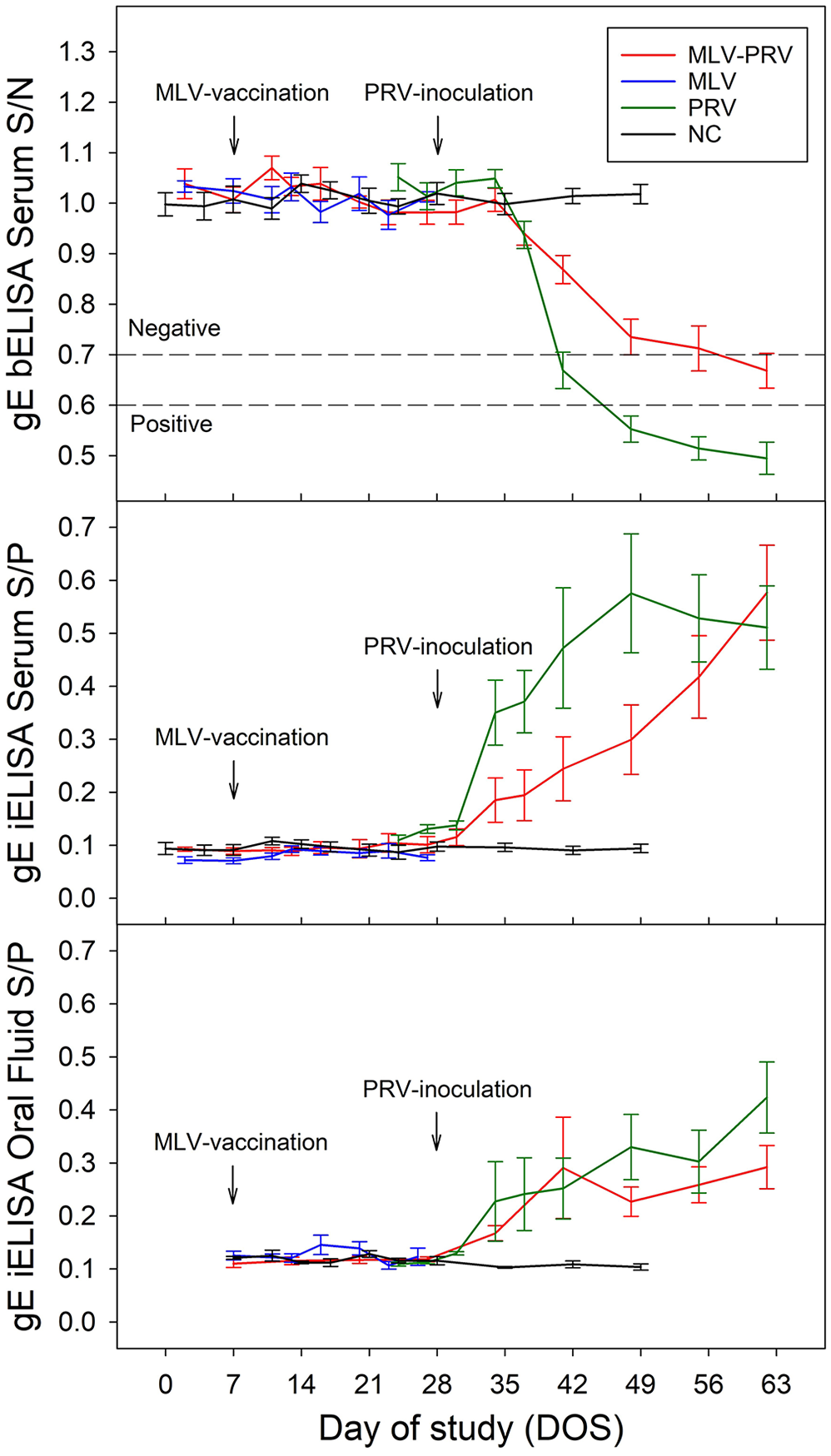
Pseudorabies virus (PRV) antibody responses (x̄ ± SE) in serum and oral fluid specimens based on a gE bELISA (Pseudorabies virus gpI antibody test kit; Idexx) and a recombinant gE iELISA testing. DOS 7: intramuscular administration of modified-live virus vaccine (Ingelvac Aujeszky MLV; Boehringer Ingelheim). DOS 28: intranasal administration of PRV isolate 3CR Ossabaw (1 × 103.5 TCID50/pig).
Using the Wilcoxon rank sum test, PRV inoculation induced a statistically significant serum iELISA S/P response in the PRV group by 14 dpi (p = 0.029) when compared to the NC group (Fig. 4), but not in the MLV-PRV group. A statistically significant oral fluid iELISA S/P response was detected by 6 dpi in both PRV (p = 0.03) and MLV-PRV (p = 0.01) groups, compared to the NC group.
Serum and oral fluid iELISA S/Ps for negative and positive samples showed distinct distributions (Fig. 5). ROC analyses of serum bELISA (n = 428), serum iELISA (n = 426), and oral fluid iELISA (n = 247) showed no significant differences in performance (bootstrap method, p > 0.05 for all pairwise comparisons; Fig. 6). The S/P cutoff between 0.1 and 0.2 resulted in the highest combination of ROC-derived diagnostic sensitivities and specificities for serum and oral fluid iELISAs (Table 2).
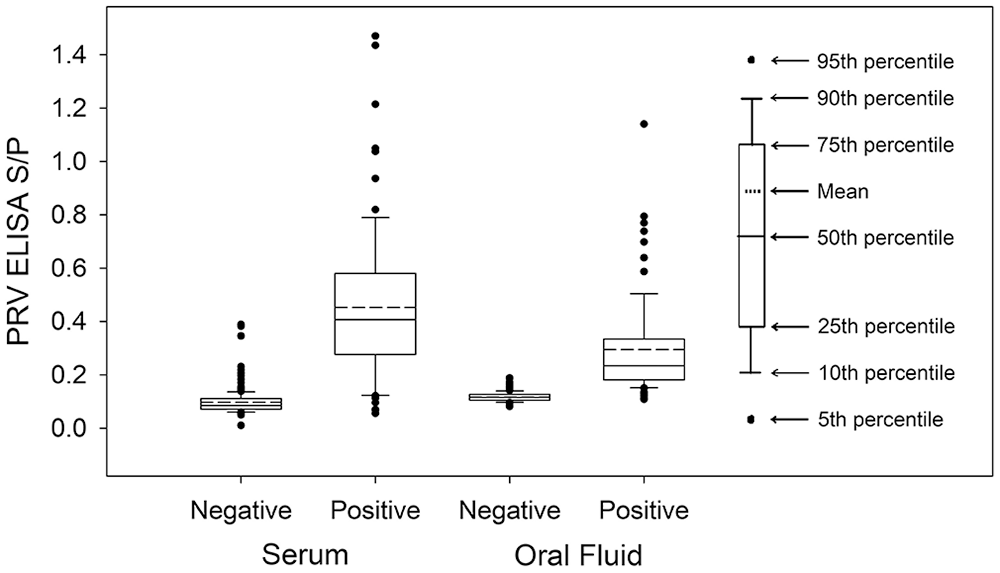
Distribution of PRV gE iELISA sample-to-positive ratio (S/P) based on testing serum (n = 381) and oral fluid (n = 225) samples from PRV-negative, PRV-vaccinated, and PRV-inoculated/-challenged pigs.
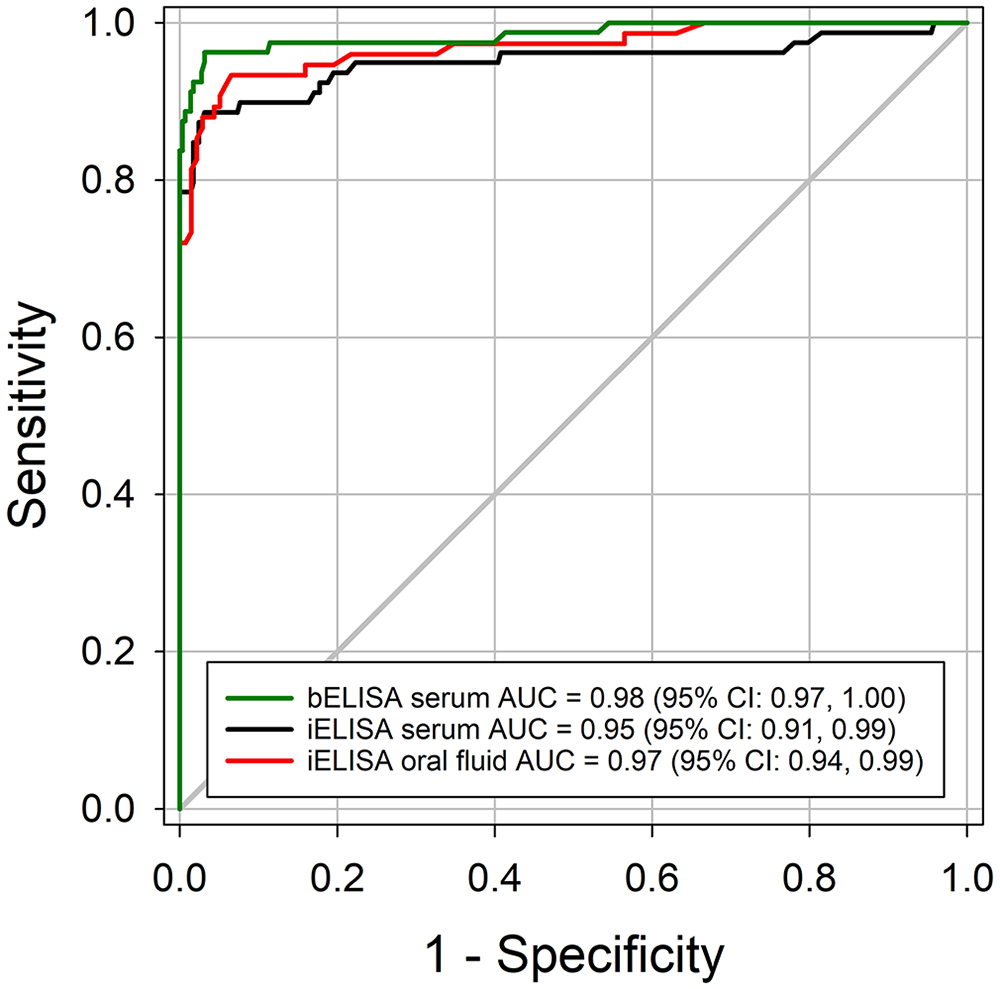
Receiver operating characteristic (ROC) curve analysis of the recombinant pseudorabies virus (PRV) gE iELISA results in swine serum and oral fluid samples. Serum and oral fluid samples collected before 7 dpi were classified as PRV gE antibody negative; samples collected after 14 dpi were considered positive to gE antibody.
Pseudorabies virus gE indirect ELISA diagnostic sensitivity (%) and specificity (%) by specimen and cutoff.
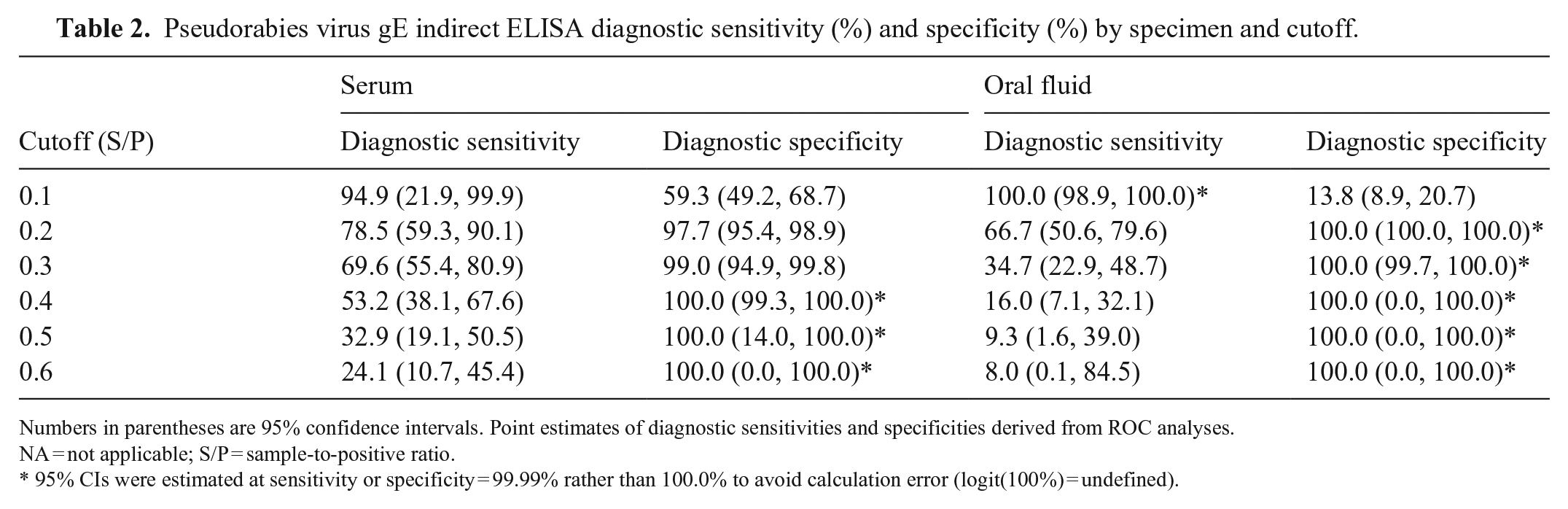
Numbers in parentheses are 95% confidence intervals. Point estimates of diagnostic sensitivities and specificities derived from ROC analyses.
NA = not applicable; S/P = sample-to-positive ratio.
95% CIs were estimated at sensitivity or specificity = 99.99% rather than 100.0% to avoid calculation error (logit(100%) = undefined).
Discussion
In comparison to PRV challenge studies, we used a moderate dose (1 × 103.5 TCID50/pig) of virus to avoid losing animals and to maximize the number of samples collected for assessment of the assays.45,48 Oral fluid samples were diluted at a lower ratio (1:1) compared to serum (1:100) to account for the lower concentration of IgG antibody in the specimen.6,26 Using samples of known PRV status, the dual-matrix gE iELISA assay detected significant gE IgG S/P responses in serum and oral fluid samples from PRV-infected pigs at 14 and 6 dpi, respectively. Consistent with previous reports of PRV serum antibody kinetics, earlier and higher serum (and oral fluid) antibody S/P responses were observed in naïve animals inoculated with wild-type virus when compared to MLV vaccinated pigs.22,46 Seroconversion was detected later by the commercial bELISA compared to the iELISA, suggesting that the serum bELISA required a higher concentration of antibodies to achieve detection. Regardless, based on ROC analyses and AUC comparisons, no difference was detected in the performance of the serum and oral fluid iELISAs and a commercial serum bELISA. Successful detection of PRV gE-specific antibody in the swine oral fluid matrix supports further exploration of the use of an oral fluid–based DIVA approach in control and eradication programs.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article, with the exception that J. Zimmerman serves as a consultant to Idexx Laboratories, on areas of diagnostic medicine independent of this study. The terms of the consulting arrangement have been reviewed and approved by Iowa State University in accordance with its conflict-of-interest policies.
Funding
Our research was supported by a grant from the Swine Health Information Center, Ames, IA, USA (grant 17-201).