
Abstract
Despite successful eradication of pseudorabies virus (PRV) from the commercial pig industry in the United States in 2004, large populations of feral swine in certain regions act as wildlife reservoirs for the virus. Given the threat of reintroduction of the virus into domestic herds, a rapid, reliable, easily implemented assay is needed for detection of PRV. Although a real-time PCR (rtPCR) assay exists, improvements in rtPCR technology and a greater understanding of the diversity of PRV strains worldwide require an assay that would be easier to implement, more cost effective, and more specific. We developed a single-tube, rapid rtPCR that is capable of detecting 10 copies of PRV glycoprotein B (gB) DNA per 20-µL total volume reaction. The assay did not produce a false-positive in samples known to be negative for the virus. The assay was negative for genetically similar herpesviruses and other porcine viruses. Our assay is a highly specific and sensitive assay that is also highly repeatable and reproducible. The assay should be a useful tool for early detection of PRV in pigs in the case of a suspected introduction or outbreak situation.
Introduction
Rapid assays are critical in the detection of diseases of economic concern in order to protect animal health and ensure food security. Around the globe, diseases such as foot-and-mouth disease, classical swine fever, African swine fever, and pseudorabies pose threats to global food security resulting from trade and commerce restrictions between disease-endemic and disease-free regions. 12 Despite the successful eradication of pseudorabies virus (PRV) from the commercial pig industry in the United States in 2004, large populations of feral swine present in at least 25 states act as wildlife reservoirs for the virus. 15 In order to rapidly detect PRV in the swine industry, a reliable molecular assay remains necessary.
PRV (also known as Aujeszky’s disease virus, or suid alphaherpesvirus 1) is a disease agent that causes global economic losses of domestic pigs each year. Although contemporary estimates are unavailable, prior to the eradication of PRV from U.S. domestic herds, the annual cost of PRV to producers was estimated to be at least US$21 million. 20 Following the successful eradication of the virus in U.S. production herds, consumers gained $336.5 million and producers gained $35.9 million in economic benefits as a result of elimination of the disease. 11
PRV can cause fatal disease in newborn pigs, respiratory disease in pigs of all ages, and reproductive failure in sows. 5 The virus is capable of causing infection in a wide spectrum of mammals; however, domestic and wild members of the Suidae family are the only hosts capable of surviving infection with the virus and can therefore serve as reservoirs for the virus. 16 Commercial swine herds are threatened with reemergence of PRV from multiple pathways, mostly notably persistently infected feral swine.7,20 Lifelong latently infected domestic and feral swine may experience an intermittent recrudescence of PRV infection, accompanied with viral shedding via oronasal or genital routes, often with low viremia.17,18,21 The virus has been also shown to remain intact in the environment from 1 to 2 wk, 20 representing a potential risk of infection to animals consuming infected carcasses. 6
PRV is a neurotropic alphaherpesvirus that is most closely related to bovine herpesvirus-1 (BoHV-1), and is monophyletic with equine herpesvirus-1 and feline herpesvirus-1. 10 Like all herpesviruses, PRV has a large genome that is ~143 kbp in length and encodes 70 genes. The envelope of the virus contains 11 glycoproteins that are targets for the host immune response. The glycoprotein B (gB) gene (also known as UL27) is conserved among PRV isolates given its role in virus penetration and cell-to-cell spread.8,17 This gene has previously been used in assays for detection of this virus.9,22
Serodetection assays are routinely used to determine the presence of PRV in feral swine, which can be as high as 38% in feral swine populations in the southern United States and Hawaii. 15 Because isolates of PRV from feral swine are presumed to be more attenuated than isolates from domestic swine,6,13,14 re-infection of domestic pigs could result in mild initial clinical signs. In that scenario, serology would have limitations as a rapid detection tool, particularly if serum was not available or animals had not yet seroconverted. Because of this threat of reintroduction of the virus in commercial herds, a rapid, reliable, easily implemented assay is needed for PRV.
Detection tests for PRV have included isolation of the virus from infected tissues, 16 or detection of antibodies to PRV by ELISA, latex agglutination, or virus neutralization tests. Following demonstration of efficacy of gene-deleted PRV vaccines, companion differential antibody ELISAs that can differentiate infected from vaccinated animals (or DIVA tests) became the gold standard for detection and differentiation between exposure to wild-type or vaccine strains of the virus.20,22 Owing to the fact that virus isolation is time-consuming and serodetection assays are not useful in infected animals that have yet to seroconvert, molecular assays are needed for the detection of PRV in commercial swine. 3 Although serologic tests provide data about PRV-specific antibody production and past exposure to the pathogen, they provide limited information about the current infection status in an animal (active vs. latent infection). 4 Factors related to the infection process (e.g., antigenicity of the infecting strain, host genetics) may delay the production of PRV-specific antibodies in an infected individual, 13 and thus a method to detect circulating virus is needed. 13 Although a previously developed real-time PCR (rtPCR) assay addressed some of these issues, the low specificity and high cost of a multiplex assay prevented its widespread use. A two primer–probe rtPCR assay that distinguishes between live-attenuated vaccinate and wild-type virus was developed nearly a decade ago9,22; however, widespread vaccination of commercial herds no longer occurs in the United States and a second probe set that detects vaccination status is not currently necessary, but would be necessary in the event of an outbreak when vaccination may be employed or in currently vaccinated herds.
We developed a rapid rtPCR that is capable of detecting 10 copies of the fragment PRV gB gene per 20-µL reaction. Both the sensitivity and specificity of the assay are high, without the need for multiple primers, probes, or reagents. The simplicity of our assay, only one primer–probe set versus a multiplex assay, likely increases reproducibility, repeatability, measurably increases specificity, and reduces the cost of reagents necessary for the assay. The assay should be a useful tool for early detection of PRV-positive animals that have not yet seroconverted. This assay is able to be used to detect infected individuals in a rapid and consistent manner.
Materials and methods
PCR development
Previously published primers 9 were redesigned (Table 1) by shortening both primers to reduce their annealing temperatures and by moving the assay probe towards the 3’-end of the forward primer (Fig. 1). Twelve sequences of the PRV gB gene in GenBank were utilized in the in silico design and analysis of these primers (Table 2). Conserved regions of the gB gene were identified by aligning the selected PRV strains in ClustalW and visualizing the output in BioEdit v.7.0 (Ibis Bioscience, Carlsbad, CA). BLASTN (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi) was used to verify the specificity of the designed primers, and the primer specification was analyzed using a multiple primer analyzer (Thermo Fisher Scientific, Waltham, MA).
Tm = temperature.

Modifications to the primer and probe oligonucleotides reported by Ma et al., 2008 9 for the detection of suid herpesvirus 1 (PRV) glycoprotein B (gB) DNA. Yellow and green highlighted areas represent the sequences of the gB primers and probe, respectively. Light gray font indicates the position of nucleotides that were previously reported by Ma et al. but were removed in the assay reported here. Bold and bright blue font indicates nucleotides that have been added to the probe and reverse primer to increase the specificity of the assay.
Nucleotide sequences and their GenBank accessions used to modify primers developed by Ma et al. 2008. 9

Sequences derived from pseudorabies outbreaks in China.
The rtPCR assay was optimized using an ultra-fast protocol (Brilliant III Ultra-Fast QPCR master mix, Agilent Technologies, La Jolla, CA) with ROX to a final reaction volume of 20 μL. The reaction contained 3.0 mm of magnesium chloride, 400 nm of forward primer gB_526F, 400 nm of reverse primer gB_613R, and 200 nm FAM-labeled gB_PRVpr probe. In order to monitor for PCR inhibitors in all sample types that would lead to false-negative results, a commercial synthetic nucleic acid internal control (VetMax Xeno internal positive control DNA, Applied Biosystems, Foster City, CA) was spiked into the PCR mixture of all reactions, along with the accompanying internal positive control assay (VIC-labeled probe and primer set; VetMax Xeno internal positive control-VIC assay, Applied Biosystems). This was performed according to the manufacturer’s instructions. Fluorescent detection was performed using a real-time thermocycler (ABI 7500 Fast rtPCR system, Applied Biosystems) and accompanying software (7500 software v.2.0.6, Applied Biosystems) using the following temperature profile: 95°C for 3 min, followed by 40 cycles of 95°C for 15 s, and 60°C for 30 s.
DNA samples
Total nucleic acids from 4 PRV-positive culture samples isolated from feral swine or a domestic dog after contact with an apparently infected feral pig were obtained for primer validation and assay optimization from the University of Florida, College of Veterinary Medicine (UFCVM; C Romero, unpublished data). Total DNA was extracted from 400 µL of infected cell culture medium from these isolates originating from animals infected in either Florida or Georgia (Table 3). The U.S. Department of Agriculture–National Veterinary Services Laboratories (NVSL) provided nucleic acids from the PRV Shope strain as well as historic field isolates from domestic pigs collected in the United States between 1982 and 2007 (n = 19). Negative controls consisted of molecular-grade water and DNA extracted from nasal swabs collected from healthy, adult domestic pigs screened for influenza viruses and West Nile virus at the UFCVM and found to be negative (D Prasko, unpublished data). To determine the specificity of the assay, NVSL provided nucleic acids from closely related herpesviruses and porcine viruses that cause respiratory and neurologic signs that include ovine herpesvirus 2 (OvHV-2), BoHV-1, bovine herpesvirus 2 (BoHV-2), and bovine herpesvirus 4 (BoHV-4). NVSL also provided nucleic acids of porcine viruses for assay exclusivity including porcine reproductive and respiratory syndrome virus (PRRSV; North American and European strains), porcine respiratory coronavirus, porcine rotavirus, porcine parvovirus, porcine teschovirus, hemagglutinating encephalomyelitis virus, and sapelovirus A.
Pseudorabies virus (PRV) isolates used to verify primer selection by conventional PCR.

To determine that the assay was capable of amplifying global PRV isolates, DNA from clinical PRV-infected animals from Italy, Belgium, Austria, Slovakia, Germany, Hungary, France, Spain, and Portugal were screened for PRV (Supplementary Table 1). A total of 64 nucleic acid samples from these countries were tested for inclusivity on the assay.
Determination of sensitivity
A conserved 100-bp region from the gB gene of the Shope strain provided by NVSL was cloned into a commercial bacterial plasmid (pCR 4-TOPO vector, Thermo Fisher Scientific). The resultant plasmid DNA clones (n = 10) were extracted (Plasmid mini kit, Qiagen, Valencia, CA), and their sequences were verified for homogeneity and parental strain. Quantitative results were based on 10-fold serial dilutions of the cloned gB fragment of the plasmid. To test assay performance, pig DNA obtained from caesarian-derived, colostrum-deprived neonates from a closed herd, free of PRV, originating from the midwestern region of the United States were used to spike with known quantities of plasmid DNA.
Determination of assay precision
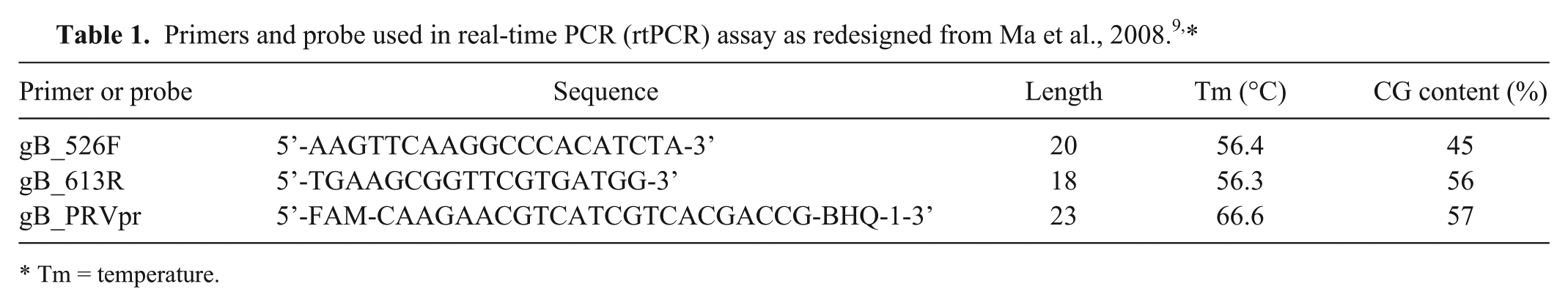
To determine assay repeatability, plasmid standards at concentrations of 107, 103, and 10 PRV gB copies/µL, representing high, medium, and low concentrations of target sequence, respectively, were run in five replicates on six individual plates. The standards were run on six different days under identical conditions. Threshold values of replicate assays within each plate were compared to determine repeatability and between plates to determine reproducibility. Here, we define repeatability (r) as the ability of a method to produce the same results from identical samples under the same conditions in the same laboratory,1-2,19 which is also known as intra-plate variability. Reproducibility (R) is defined as the ability of a method to produce the same values for replicate samples run under the same conditions by different users on different days, which is also known as inter-plate variability. Because analyses took place in a single laboratory, identical assays were repeated on different days, on different plates by three different users. Reproducibility and repeatability were calculated using the standard normal cumulative distribution function in SAS (SAS Institute, Cary, NC) using 30 replicates.1-2,19 The r or R values indicated the maximum difference among replicate quantification cycle (Cq) values on a log10 scale allowable within a 95% probability framework (α = 0.05). We used multiple technicians and machinery commonly used at animal health laboratories in order to facilitate transferability of the assay.
Results
rtPCR specificity and sensitivity
The specificity of the assay was determined by testing genomic DNA from closely related viral pathogens, uninfected domestic pigs, and global and domestic isolates of PRV. The assay was highly specific and did not result in false-positives when testing any of the uninfected pig samples or other viruses tested; 25 of 25 samples, including domestic pig samples, were negative for PRV. The assay initially detected all but 1 of 64 geographically diverse isolates of PRV. A single sample failed to amplify (Spain 1258-3094) and exhibited the presence of inhibitors, which was identified by examining the amplification plot (7500 software v.2.0.6, Applied Biosystems) for the internal positive control (VetMax Xeno, Applied Biosystems). The sample was successfully amplified after minimizing the effect of the inhibitors by diluting (1:10) the extracted nucleic acids with molecular-grade water. All 23 domestic PRV isolates provided by either NVSL (n = 19) or UFCVM (n = 4) amplified in the assay. In total, PRV DNA was detected in 87 of 87 (100%) domestic and international isolates tested. PRV DNA was not detected in any of the PRV-negative samples or non-template controls.
The limit of detection of the assay was 10 copies of target sequence per 20-μL reaction of PRV. The sensitivity of the assay was not significantly different in the presence of DNA from pigs. Using water as diluent, the average Cq for the detection of 10 copies of PRV DNA was 34.8. In the presence of domestic pig DNA, 10 copies of PRV (representing the lower limit of detection of the assay) were detected in 10 replicates with an average Cq of 36.2, indicating minimal loss of sensitivity.
Assay precision
Repeatability (r) values representing the maximum expected difference between rtPCR replicates (with 95% probability) were obtained for each of three template quantities. For the highest concentration of template (107 copies/µL), r = 0.6 and R = 0.9 were estimated. At medium template concentration (103 copies/µL), r = 0.4, R = 1.6; and at the lowest concentration (10 copies/μL), r = 0.8 and R = 1.9. The r values were similar at all 3 concentrations, making it unnecessary to reduce the cutoff threshold of the assay. As expected, the assay was less repeatable at the lowest end of the range (10 copies). Overall, at a high concentration, we found that replicates did not vary more than 0.6 Cq and did not vary more than 0.4 and 0.8 Cq at medium and low template concentrations, respectively. The smallest r value was obtained for 1,000 copies, as replicates were nearly identical at this concentration. However, replicates at all concentrations performed well on this assay (Fig. 2a). Between users (n = 3), values did not vary by more than 2 Cq values between plates, run on different days (range of R = 0.9–1.9). The variance between plates was 7.0–8.7% (data not shown), which is within the range of acceptability (<10%) as minimal variation was observed between runs (Fig. 2b). 19
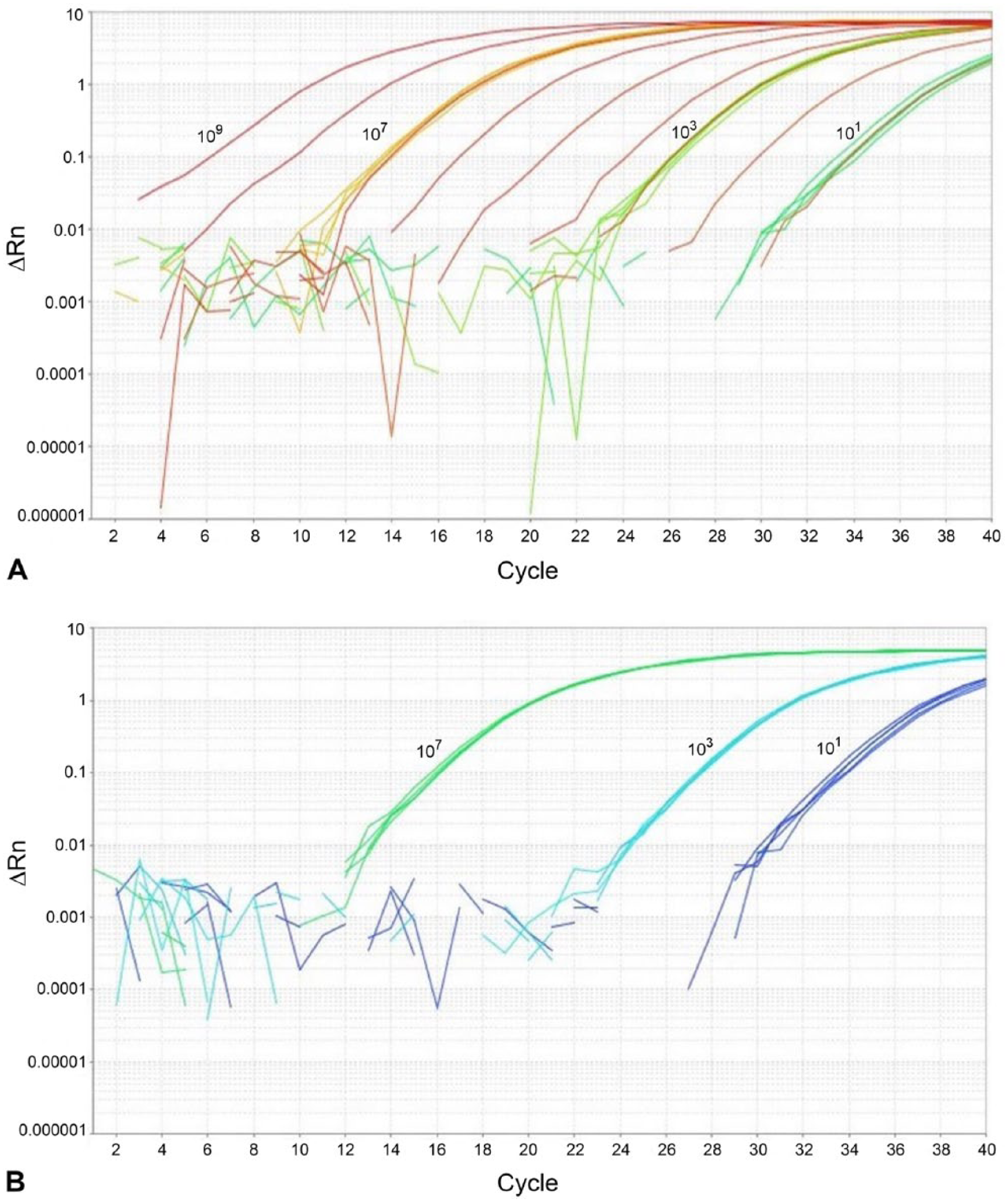
Assay performance was determined by running five replicates of plasmid standards six times on individual plates, run on different days. Dilutions of 107, 103, and 10 copies of target DNA were utilized to determined assay precision.
Discussion
Our rtPCR assay exhibited a higher specificity than the previously published assay (64–76% in the previous assay vs. 99% in this assay). The revised assay was able to detect the virus in multiple sample types (culture isolates, nasal swabs, tissues homogenates) collected from PRV-infected wild pigs from Europe and feral swine from the United States (unpublished data); the assay did not detect PRV false-positives in any of the DNA samples collected from uninfected domestic pigs, other porcine viruses, or phylogenetically related herpesviruses. The use of the internal positive control DNA prevented a false-negative resulting from PCR inhibition, which has the potential to arise from degraded template DNA or inhibitors present in the sample from the host or the environment.
There is a PCR for the gE gene that is currently available that could be used in the event of an outbreak where vaccination was used in a herd. 9 The simplicity of our newly developed assay, using only one primer–probe set for the detection of PRV DNA, likely increases reproducibility, repeatability, and reduces the cost of reagents necessary for the assay. An optimal internal positive control can be used for low template quality or quantity inputs, but may not be necessary in all scenarios. This updated rtPCR assay could be used as a molecular tool in conjunction with serologic methods as part of a surveillance program for detecting diverse wild-type PRV strains distributed worldwide. It is capable of differentiating PRV from herpesviruses that infect other hoof stock. The sensitivity of our rtPCR assay facilitates the early detection of PRV-infected domestic swine as the result of a pathogen introduction or outbreak, playing a pivotal role in supporting PRV surveillance and preventing widespread introduction in commercial swine herds across the United States.
Footnotes
Acknowledgements
We thank all of the researchers who provided isolates or DNA samples of PRV, including C Romero at the University of Florida, College of Veterinary Medicine. We thank D Prasko and S White at the University of Florida for providing nasal swabs obtained from domestic swine. We also thank K Shanmuganatham, R Tell, and J Schiltz for valuable comments on this manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This material was made possible, in part, by a Cooperative Agreement from the United States Department of Agriculture’s Animal and Plant Health Inspection Service (APHIS). It may not necessarily express the views of APHIS.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.