
Abstract
Acute bovine liver disease (ABLD) is a sporadic hepatic disease affecting cattle in southern Australia, characterized histologically by striking periportal hepatocellular necrosis. The cause of ABLD is unknown; however, the seasonality and acute presentation of outbreaks suggest mycotoxin involvement. We describe here the clinical and pathologic findings of ABLD in 45 naturally affected cattle from 13 outbreaks occurring from 2010 to 2019 in Victoria, Australia. Outbreaks occurred in herds located along the southern coastal plain of Victoria and were observed most frequently in lactating dairy cattle. Clinical signs commonly included a combination of mild photosensitization, progressive neurologic signs, and hypogalactia, which preceded death by ≤ 48 h. All affected animals had marked elevations in activities of glutamate dehydrogenase, aspartate aminotransferase, and gamma-glutamyl transferase. At autopsy, the most common lesions were serosal petechiae and/or gastrointestinal hemorrhage, and hepatomegaly with a pronounced hepatic reticular pattern. The principal histologic lesion was widespread—severe periportal hepatocellular coagulative necrosis and erythrocyte pooling—which often extended to massive necrosis. Lesions in other organs were uncommon. Our study of ABLD suggests involvement of a potent hepatotoxin that is either directly cytopathic or requires bioactivation by periportal-specific enzymes.
Acute bovine liver disease (ABLD) is a hepatic disease that mainly affects grazing cattle in the high rainfall regions of southeastern Australia; primarily Victoria, Tasmania, and southeast South Australia, with rare reports outside of these areas.8,13,20 To our knowledge, there are currently no reports of ABLD outside Australia. ABLD occurs commonly in the days to weeks following > 4 mm rainfall, with high humidity, calm conditions, and a temperature of > 12°C. 20 There is no apparent age, sex, or breed predilection; however, reports are most common in lactating dairy cattle, particularly those managed with a rotational grazing pattern.16,20 ABLD is highly seasonal, with outbreaks occurring in autumn and winter, and occasional outbreaks in spring, when climatic conditions support heavy dews and warm days.16,20 The disease is sporadic and unpredictable, with highly varied morbidity and mortality rates.8,13
ABLD has been recognized since the 1950s; however, many aspects of the disease remain elusive. 20 The cause of ABLD is unknown but is generally accepted to be a toxin. The acute clinical presentation and seasonality associated with ABLD are suggestive of mycotoxicosis.16,20 Notably, outbreaks are often associated with the presence of dry, senescent Cynosurus echinatus (rough dog’s-tail grass) from previous seasons, and the emergence of lush green pastures. 16 Although toxicity of the grass itself has not been demonstrated,1,16 the grass has been identified as a risk factor by state departments given its consistent presence at the time of outbreaks. Other risk factors include lactation and fodder scarcity. 16 Current investigations are focused on Pyrenophora (previously Drechslera) spp. of fungi, which have been isolated consistently from C. echinatus at the time of outbreaks.1,16 Difficulties culturing P. siccans in significant quantities have limited studies into the effects of P. biseptata, with in vitro and in vivo studies unable to prove the toxicity of this species to date.1,16
Microscopically, liver lesions of ABLD are recognized by striking periportal-to-massive hepatocellular necrosis. 16 Periportal necrosis is an uncommon lesion in cases of plant and fungal-associated hepatotoxicosis. In Australia, this distribution of hepatic necrosis is more commonly caused by Myoporaceae toxicity, but access to these plants has been excluded in all cases of ABLD to date. 20 Diagnostic criteria for ABLD, to date, are limited to the presence of periportal or massive hepatic necrosis and exclusion of other hepatotoxins. 20 Myoporaceae toxicity is excluded through paddock examination; cases of massive necrosis require additional exclusion of cyanobacterial (Cyanophyceae, blue-green algae) toxicity through examination of water sources.5,20 In southern Australia, causes of acute hepatotoxicosis, including Xanthium strumarium, Cestrum parqui, acute pyrrolizidine alkaloidosis, acute lupinosis, Brassica-associated liver disease, and less commonly, punicalagin or amatoxin toxicosis, are excluded on the basis of histopathologic features. 17 Other causes of acute hepatotoxicosis, such as Lophotoma spp. (sawfly) larvae, Cycad spp., and Lantana camara, are excluded on the basis of histopathologic features and geographic distribution.5,17
ABLD is recognized by veterinarians and pathologists working in endemic areas of Australia; however, studies characterizing clinicopathologic, gross, and microscopic features of this disease are not present in the literature. Here, we systematically describe the spectrum of clinical presentations and hematologic, biochemical, and pathologic findings observed in a large number of cattle naturally affected by ABLD in 13 outbreaks across Victoria, Australia.
Materials and methods
Case selection
We included cases from 13 ABLD outbreaks occurring between 2010 and 2019 across 11 properties in southern Victoria, ranging from the Warrnambool region in the west to Wellington in the east (Fig. 1). For inclusion in the study, a diagnosis of acute hepatotoxicity consistent with ABLD was made by veterinary pathologists employed by the Centre for AgriBioscience (Victoria, Australia). All cases had periportal hepatic necrosis and thus did not require clinical exclusion of cyanobacterial toxicity. Paddock examination to exclude Myoporaceae toxicity was performed in all cases (Suppl. Table 1). Samples with preanalytic artefact, including hemolysis indices of 3 or 4, were excluded. We included 45 affected individuals: blood samples from 27 cases and histopathology submissions inclusive of liver from 23 cases; 5 cases had both blood and liver samples examined. Of the histopathology cases, 18 (78%) were submitted with a description and/or photographs of gross postmortem findings by the submitting veterinarian. Clinical pathology samples that were not submitted with tissue samples were included, on the basis of being from the same outbreak and having the same clinical presentation as cattle for which a histologic diagnosis was made. Routine postmortem examination and sample collection were performed by private veterinarians or veterinarians employed by the Department of Environment and Primary Industries, Victoria.
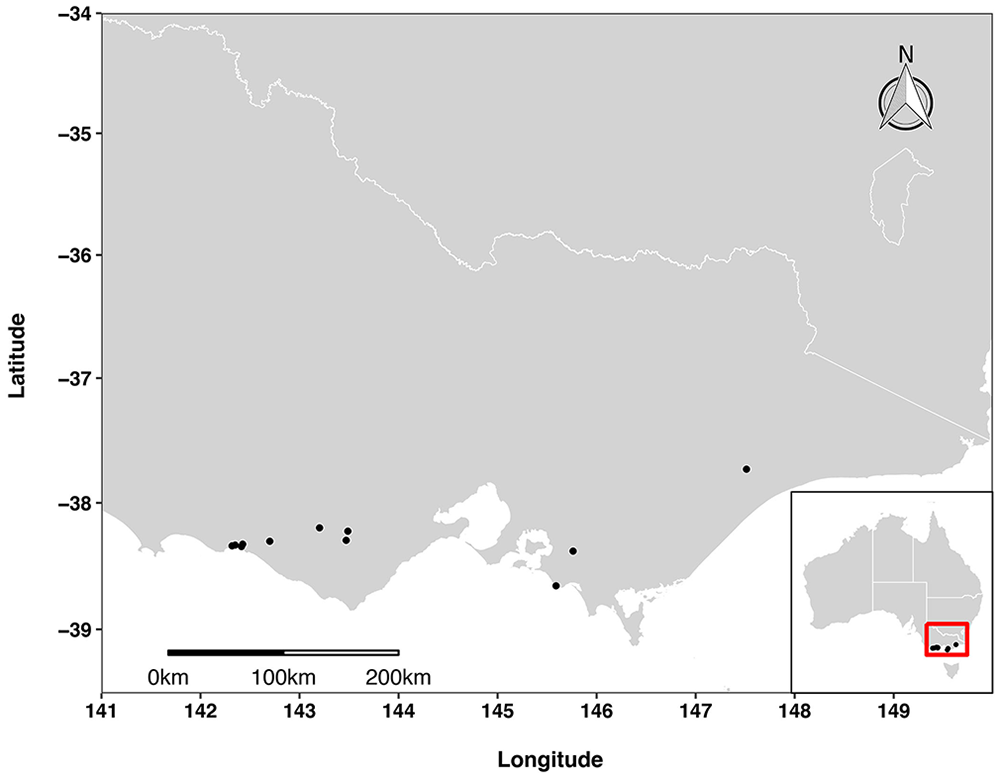
Geographic distribution of 13 outbreaks of acute bovine liver disease on 11 properties in Victoria, Australia, 2010–2019.
The number and types of tissue submitted with liver (n = 23) varied by case, but included kidney (14), myocardium (10), lung (9), spleen (9), gastrointestinal tract (7), and brain (6). Gastrointestinal tract specimens included rumen (4), reticulum (2), omasum (1), abomasum (5), small intestine (7), ileocecal valve (1), cecum (1), and colon (5). Infrequently collected specimens included cervical spinal cord (2), skin (1), bone marrow (1), mesentery (1), lymph node (1), and adrenal gland (1).
Examination of whole blood, serum, and tissues
All postmortem specimens were collected and fixed in 10% neutral-buffered formalin for at least 24 h prior to processing. Fixed tissues were processed routinely to paraffin wax and 5-µm sections prepared for examination. All sections were stained with hematoxylin and eosin (H&E) for light microscopy; additional special stains Perl Prussian blue, Martius scarlet blue (MSB) trichrome, and Masson trichrome were used to highlight features such as iron, fibrin, and collagen, respectively. Whole blood and serum samples were processed at 2 separate laboratories; hematology was performed for 13 cases, and biochemistry was performed for 27 cases. Clinicopathologic data were divided into 2 groups based on the reference intervals (RIs) used (Table 1; Suppl. Table 2). Group 1 included cases analyzed at the Centre for AgriBioscience, including 17 biochemistry panels (Randox reagents; BS-200 chemistry analyzer; Mindray) and 4 hematology panels (Vetscan HM5; Abaxis) with differential white cell counts carried out by manual smear assessment. Group 2 included cases analyzed at a private laboratory, including 10 biochemistry panels (Advia 1800; Siemens) and 9 hematology panels (Cell-Dyn 3700; Abbott).
Hematologic and biochemical findings in cattle affected by acute bovine liver disease, grouped by reference laboratory.
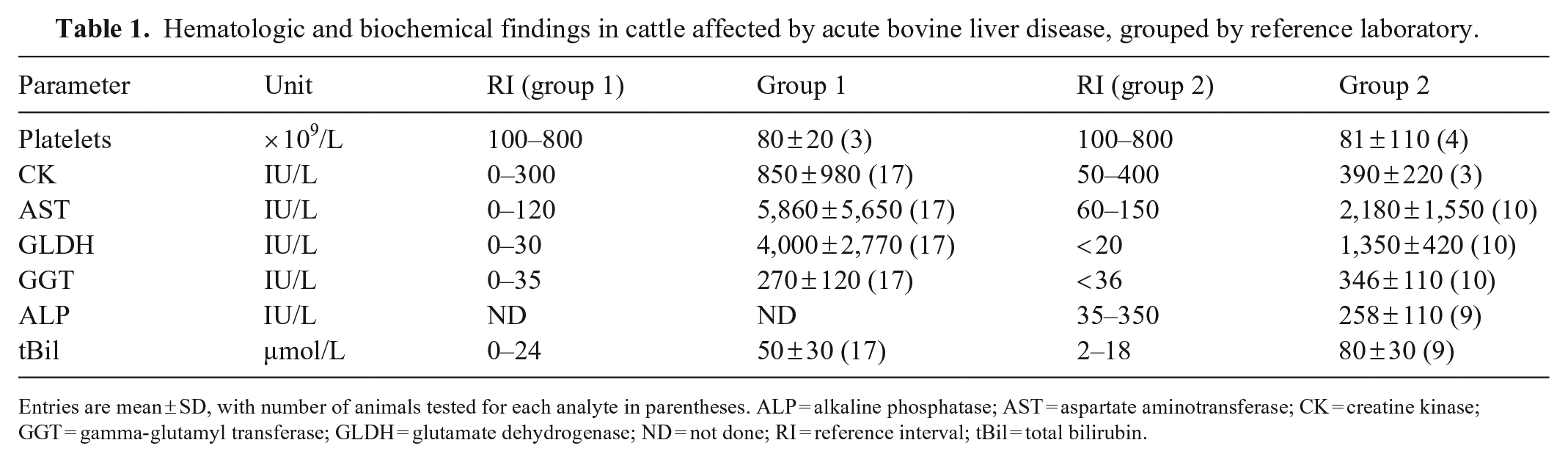
Entries are mean ± SD, with number of animals tested for each analyte in parentheses. ALP = alkaline phosphatase; AST = aspartate aminotransferase; CK = creatine kinase; GGT = gamma-glutamyl transferase; GLDH = glutamate dehydrogenase; ND = not done; RI = reference interval; tBil = total bilirubin.
For all liver specimens, ≥ 20 hepatic lobules were assessed. The portal tracts were assessed for ductular reaction and fibrosis. The hepatic parenchyma was assessed for reversible or irreversible hepatocellular injury, the location of injury, and the percentage of the lobule affected; pigment deposition within hepatocytes or Kupffer cells; cholestasis within canaliculi; and vascular changes within central and portal veins. Both the portal and parenchymal compartments were assessed for infiltration of inflammatory cells, neoplastic cells, evidence of preexisting hepatic injury, and evidence of other toxic or infectious agents. Histologic grading (Suppl. Table 3) and description of lesions was performed without knowledge of the clinical details of each case. Necrosis was defined as including one or more of the following features: nuclear loss, pyknosis, karyorrhexis or karyolysis, increased cytoplasmic eosinophilia or pallor, and architectural dissociation. Reversible cell injury was defined as feathery or hydropic degeneration. The character and severity of inflammation was noted; however, grading was complicated given the often-marked pooling of erythrocytes in examined areas. Fibrosis was characterized. 9 Features such as biliary mucostasis and erythrophagocytosis were widespread and not classified on the basis of severity. Features such as megalocytosis, pigment deposition, and cholestasis were either seen infrequently or not at all and were not considered major pathologic processes.
Results
Outbreak distribution
All outbreaks occurred along the coastal plain of Victoria (Fig. 1), south of the Great Dividing Range, within 55 km of the coastline and between 12 and 134 m above sea-level. This is a cool temperate region, with the mildest climate of all Victorian regions. Summers are mild-to-warm, with the hottest temperatures in January and February, and winters are mild-to-cool, coldest in July. Rainfall is greatest during winter, although summer rainfall may be heavy. The coastal plain is generally wetter than other parts of Victoria, except for areas to the west of Melbourne, where annual rainfall may be < 600 mm. Land use in the southern coastal plain is dominated by primary production and conservation reserves.
Clinical presentation
Among the 45 individual animals included in our study, 41 of 45 (91%) were > 2 y old; 4 (9%) were 16 mo old; 44 were female, 1 was male (Table 2). Thirty-eight of 45 (84%) cases were Holstein Friesians, all of which were mid-lactation at the time of outbreak, and the remaining 7 of 45 (15%) were Angus cattle, including the 4 heifers, 1 steer, and 2 cows with unspecified lactation status (Table 2). The duration of clinical signs prior to death or euthanasia was reported in 23 of 45 cases (Suppl. Table 4). Of these, 18 of 23 (78%) had clinical signs for < 48 h prior to death or euthanasia, including 11 of 23 (48%) cases that had clinical signs for < 24 h prior to death or euthanasia. Two individuals (9%), cases 24 and 25, had clinical signs for 22 and 23 d, respectively, and were euthanized because of chronic ill-thrift and hypogalactia (case 24) and severe photosensitization (case 25). In 17 cases, clinical history allowed determination of the time on a new paddock prior to the onset of clinical signs, allowing estimation of the time from potential first exposure to clinical disease: 3 of 17 individuals (18%) developed clinical signs within 24 h of movement onto a new paddock, 6 of 17 individuals (35%) developed signs 24–48 h after introduction, 8 of 17 individuals (47%) within 48–72 h of introduction, with an average of 55 h (Suppl. Table 4). The mean morbidity and mortality rates between the 13 outbreaks were 21.3% and 5.5%, respectively, but varied between outbreaks (Table 2). Outbreaks were observed most frequently throughout autumn (8 of 13 [61%]) and early winter (3 of 13 [23%]) and rarely in mid-winter (1 of 13 [8%]) and spring (1 of 13 [8%]; Table 2).
Morbidity, case fatality, and mortality rates for outbreaks of acute bovine liver disease, including property and case identification, date of outbreak, and signalment of affected animals.
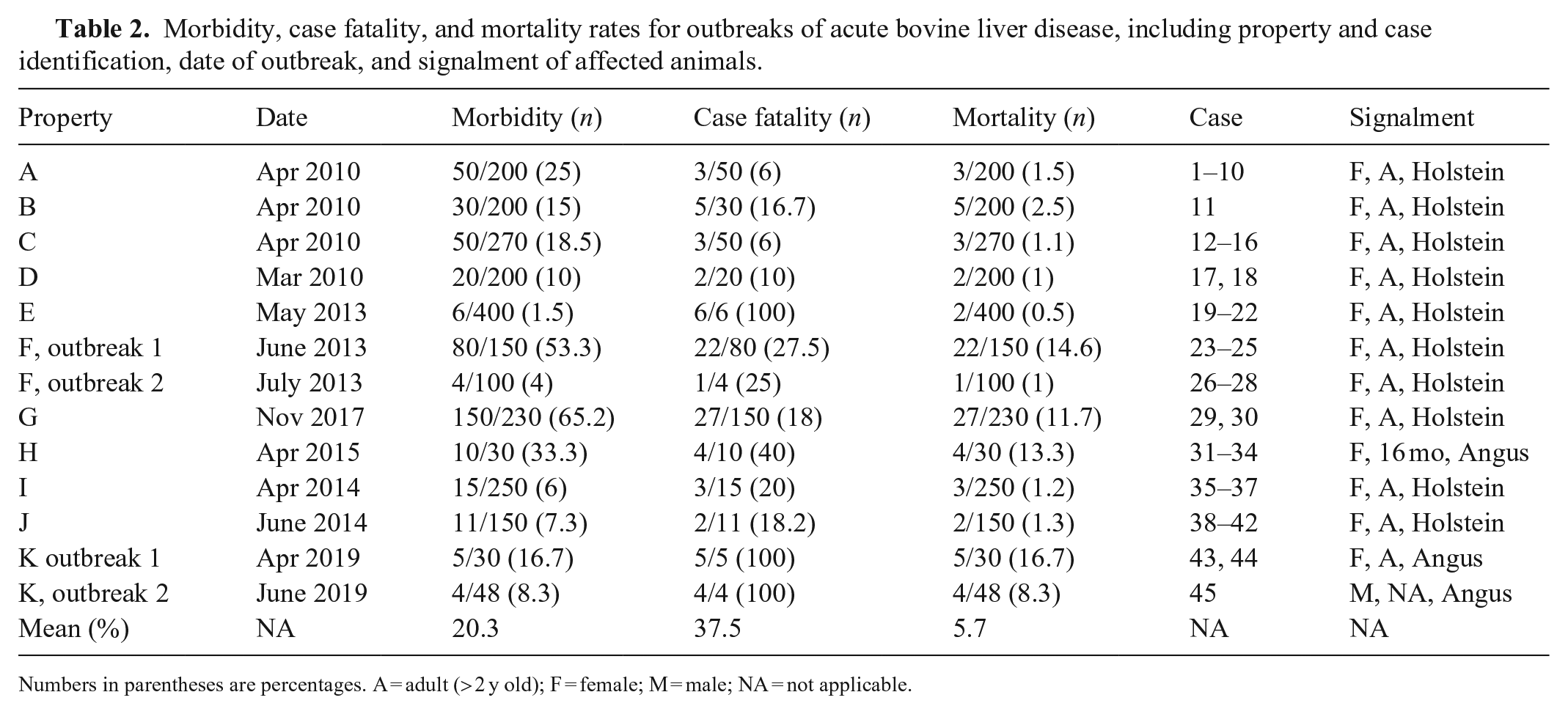
Numbers in parentheses are percentages. A = adult (> 2 y old); F = female; M = male; NA = not applicable.
The most common clinical signs (Suppl. Table 4) included photosensitization (23 of 45 [51%]), recumbency (20 of 45 [44%]), progressive neurologic signs (17 of 45 [38%]), hypogalactia (12 of 38 [32%]), and jaundice (14 of 45 [31%]). Photosensitization was often mild, characterized by erythema of the periocular skin, vulva, and udder, with some exudative lesions. Severe photosensitization was observed only in case 25. The second chronically affected animal (case 24) was euthanized because of sudden hypogalactia but showed no other clinical signs at the time of euthanasia. Neurologic signs were invariably progressive and included dullness, paresis, reduced responsiveness, ataxia, agitation, aggression, hyperesthesia, and vocalization, and on one property was seen to progress to terminal convulsions. Less common clinical signs (Suppl. Table 4) included mild epistaxis, hematuria, and rectal hemorrhage. A number of animals had gastrointestinal signs, including constipation (9 of 45 [20%]), bloat (7 of 45 [16%]), and ruminal stasis (5 of 45 [11%]). Vital signs were variably reported, including heart rates (n = 16) of 66–140 beats per minute, and respiratory rates (n = 4) that were noted as mildly elevated in all cases. Temperature (n = 13) was reported as hypothermia (n = 5), pyrexia (n = 2), 38°C (n = 2), and 37.9–39.9°C for all animals in a single outbreak (n = 4).
Laboratory findings
Hematology and biochemistry (Table 1; Suppl. Table 2) revealed marked elevations in glutamate dehydrogenase (GLDH) activity in all 27 cases, accompanied by marked, but less striking, elevations in gamma-glutamyl transferase (GGT) activity. Marked elevations in aspartate aminotransferase (AST) activity were present in all cases and occurred with concurrent creatine kinase (CK) elevations in 16 of 19 (84%) cases. Hyperbilirubinemia was common and varied in severity; however, most cases had moderate-to-marked elevations. Leukocyte changes were variable and mild; the most common finding was mild-to-moderate neutrophilia, often insufficient to cause leukocytosis, sometimes accompanied by mild lymphopenia and/or a left shift. Thrombocytopenia was present in 6 of 7 (86%) cases and was mild-to-marked. All other findings were either mild or nonspecific, including occasional mild azotemia and mild hyperproteinemia. Mineral and electrolyte abnormalities were variably present; hypocalcemia (1.96–1.14 mmol/L, RI: 2.0–2.75 mmol/L) was the most common derangement, seen in 8 of 16 (50%) cases.
Gross and microscopic findings
Liver
Macroscopic abnormalities (Suppl. Table 1) were present in 9 of 18 (50%) cases, characterized by hepatomegaly (7 of 18 [39%]) and/or a prominent reticular pattern (6 of 18 [33%]; Fig. 2). Jaundice was another frequent finding, seen in 8 of 18 (44%) cases (Suppl. Table 5). Rare incidental lesions, including fibrosis and fibrous adhesions, were not associated with a longer reported clinical time course and were presumed to be preexisting.
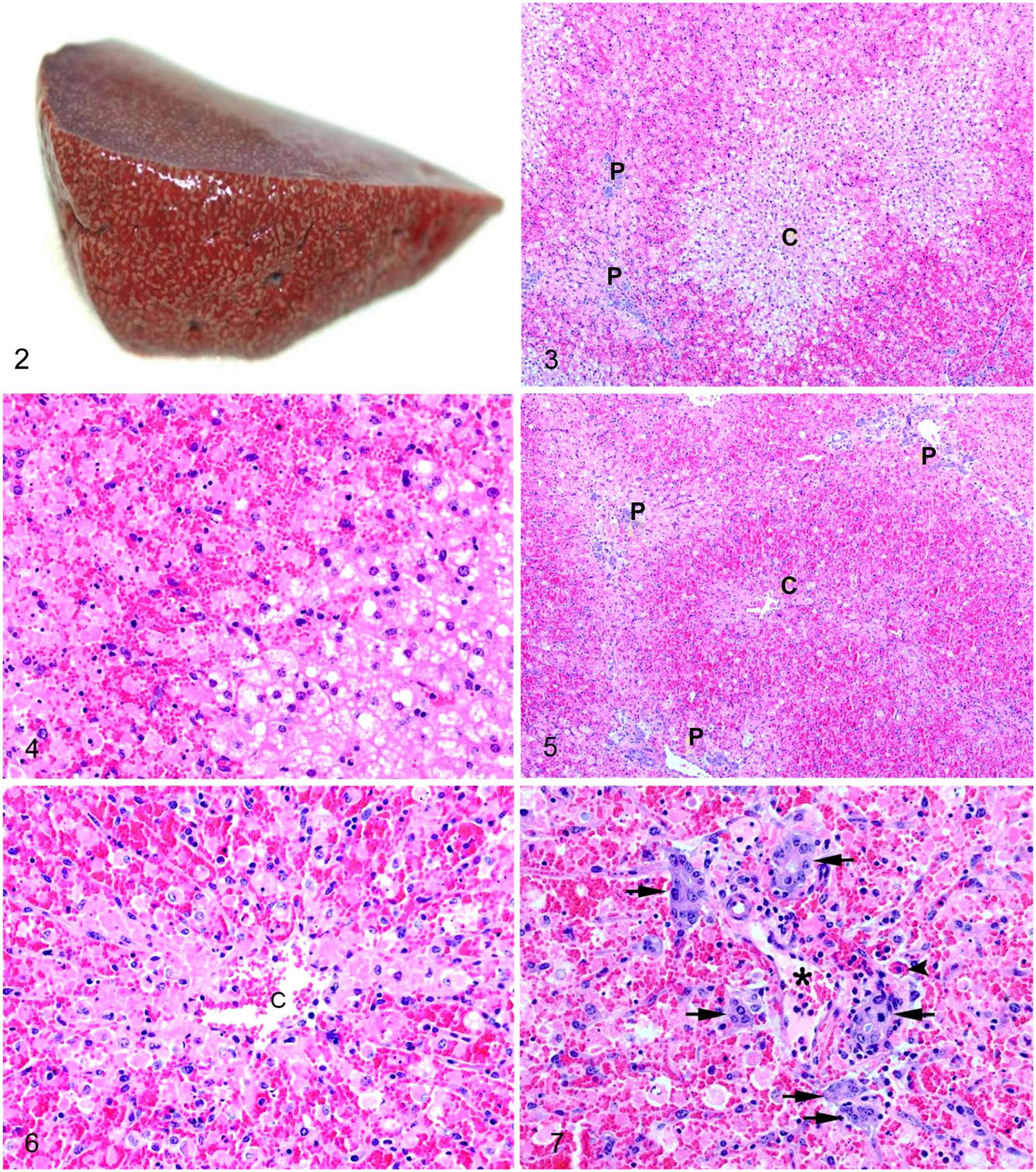
Acute bovine liver disease. C = central vein; P = portal tract.
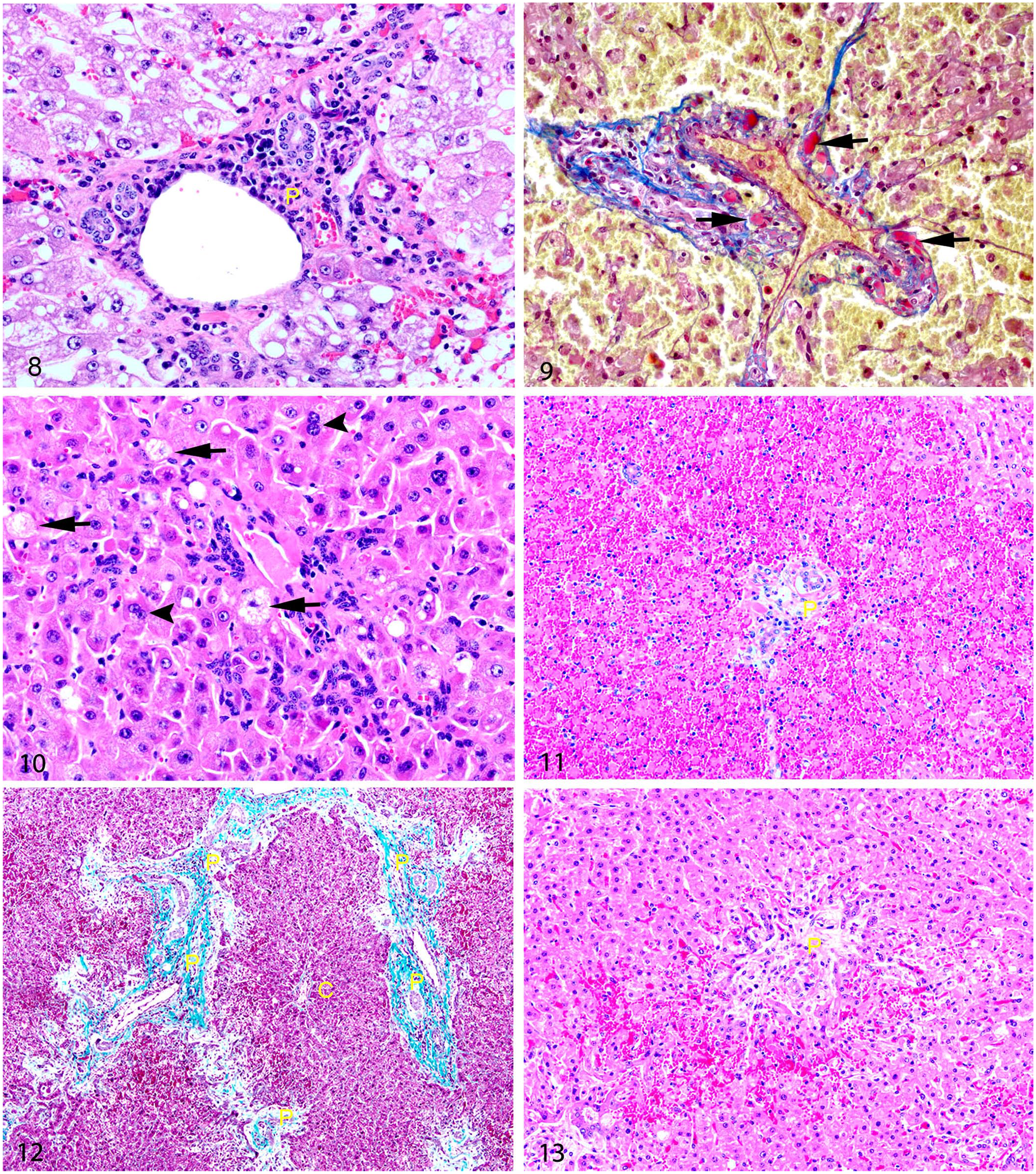
The liver was examined microscopically in 23 cases; zonal hepatocellular necrosis was observed in all cases; in 3 of 23 (13%) cases, necrosis was confined to the periportal zone (zone 1), in 11 of 23 (48%) cases necrosis extended into the midzone (zones 1 and 2; Fig. 3), and in 9 of 23 (39%) cases, there was massive necrosis, most advanced in the periportal regions (Figs. 5, 6). In all cases, necrosis was coagulative, characterized by retention of architecture and cellular outlines (Figs. 4, 6). Severe necrosis, affecting > 50% of the lobule (Fig. 5), was present in 15 of 23 (65%) of cases; necrosis was moderate in 5 of 23 (22%), and mild in 3 of 23 (13%) cases (Suppl. Table 1). In all cases, remaining hepatocytes had hydropic degeneration (Figs. 3, 4). Hemorrhage was a notable feature in all acute cases, characterized by pooling of erythrocytes in necrotic areas with occasional erythrophagocytosis by macrophages (Fig. 7). Hemosiderin-laden macrophages were rare or absent in all cases.
Ductular reaction (DR) was present in 22 of 23 (96%) cases (Fig. 7); 13 cases had mild DR, and 9 cases had moderate DR (Suppl. Table 1). Fibrosis was absent in 18 of 23 (78%) cases and was mild in the remaining 5 (22%). Inflammation was most often characterized by mild-to-moderate infiltration of neutrophils and fewer macrophages into areas of necrosis; this was observed in all clinically acute cases (21 of 23 [91%]). The 2 chronic cases (cases 24 and 25) had moderate infiltration of lymphocytes and plasma cells into the portal and periportal regions (Fig. 8). Mild lymphoplasmacytic portal inflammation was also seen in cases 19 and 36. Mild vascular changes were seen in 21 of 23 (91%) cases, characterized by endothelial hypertrophy (Fig. 7) primarily affecting portal veins (14 of 21 [67%]) or both portal and central veins (7 of 21 [33%]). Endothelial loss and attenuation affecting central veins was seen in 4 of 23 (17%) cases, accompanied by hydropic degeneration in perivenular hepatocytes. Intravascular fibrin thrombi (Fig. 9) were present in 12 of 23 (52%) cases and were located in portal regions (5 of 12 [42%]), margins of necrosis (1 of 12 [8%]), or both (6 of 12 [50%]). Six (50%) cases had gross evidence of hemorrhagic diathesis at postmortem (cases 1, 9, 20, 30, 31, 45). Cases 35, 42, and 45 did not have evidence of hemorrhagic diathesis; cases 10, 18, and 26 had no available data for macroscopic findings.
Acute bovine liver disease. C = central vein; P = portal tract.
Lesion variation in ABLD-affected livers
Cases 24 and 25 survived 22 and 23 d, respectively, after an initial ABLD outbreak. Both had periportal to occasionally midzonal cell swelling and microvesicular cytoplasmic vacuolation affecting 20–30% of hepatocytes (Fig. 8). Occasional small foci of hepatocellular coagulative necrosis were seen in periportal regions, affecting < 5% of hepatocytes. Hepatocytes within the midzonal and periportal zones had mild anisocytosis and anisokaryosis with occasional megalocytosis, occasional binucleation, and rare trinucleation (Fig. 10). Both cases also had mild-to-moderate portal-to-periportal lymphoplasmacytic infiltration. Endothelial hypertrophy affecting portal veins was seen in both cases. Fibrosis was mild in case 24 and absent in case 25, and ductular reaction was mild in case 24 and moderate in case 25 (Suppl. Table 1).
Further variation in histopathology was seen in case 44, in which lesion severity varied between liver lobes; grossly, the left liver lobe was shrunken and contained pale, firm areas consistent with fibrosis; the remainder of the liver was diffusely swollen. Microscopically, the left liver lobe had moderate diffuse ductular reaction, portal fibrosis, and mild periportal necrosis (Figs. 12, 13). In contrast, the swollen regions were affected by diffuse, marked necrosis of periportal-to-midzonal hepatocytes, also extending into centrilobular regions (Fig. 11).
Gall bladder
Macroscopic gall bladder lesions (Suppl. Table 1) were seen in 3 of 18 (17%) cases, including edema of the gall bladder wall (2 of 18 [11%]) or mucosal hemorrhage (1 of 18 [5%]). Histologic examination of the 3 abnormal gall bladders and an additional grossly unremarkable case, revealed severe submucosal edema, congestion, and multifocal hemorrhages in all cases. Case 23 also had marked lymphocytic cuffing of submucosal blood vessels. Mucosa was absent in all cases (postmortem artefact).
Kidneys
Kidneys were collected from 14 individuals. In cases 23 and 45, there was macroscopically mild swelling and pallor; in all others, no macroscopic abnormalities were noted (Suppl. Table 5). Microscopic examination of kidneys from case 23 revealed no histologic changes. Histologic renal lesions were evident in 4 individuals: cases 20, 41, 42, 45. Case 20 had moderate cytoplasmic vacuolation of tubular epithelium at the corticomedullary junction, consistent with reversible cell injury. Cases 41 and 42 had mild, focally extensive tubular epithelial necrosis affecting both the proximal and distal convoluted tubules. Case 45 had random single-cell necrosis of both proximal and distal convoluted tubular epithelium.
Lungs
In all cases, lungs were grossly unremarkable. Of 9 cases examined microscopically, 3 animals had moderate patchy emphysema and/or moderate patchy atelectasis, considered agonal change.
Gastrointestinal tract
Free luminal gastrointestinal hemorrhage was observed macroscopically in 5 of 18 (28%) cases, affecting the rumen (1), abomasum (4), small intestine (2), and large intestine (3; Suppl. Table 5). Sections of gastrointestinal tract were examined microscopically from 6 animals only, 1 of which (case 41) had grossly evident hemorrhage in the abomasum and large intestine; in this case, histopathology of the large intestine revealed multifocal superficial mucosal hemorrhages in the absence of necrosis. All other animals had no significant microscopic lesions.
Spleen
The spleen was collected from 9 animals; 5 had no microscopic findings; 4 (cows 19, 20, 25, 35) had numerous hemosiderin-laden macrophages.
Serosae
Serosal hemorrhage was present macroscopically in 9 of 18 (50%) cases, seen primarily as petechiae and less commonly ecchymoses (Suppl. Table 5). This included epicardial (7 of 18 [39%]), endocardial (5 of 18 [28%]), pericardial (4 of 18 [22%]), and abdominal serosal (5 of 18 [25%]) hemorrhage. Other sites of hemorrhage were also noted, including petechiation and ecchymoses of the subcutaneous tissues in 1 case and skeletal muscle in a further 2 (10%) cases.
Discussion
The sporadic nature of ABLD makes accumulation of large numbers of cases difficult; the collection of distinct outbreaks in Victorian cattle properties in Australia over a 9-y period allowed us a unique opportunity to study the clinical and pathologic features of this disease. There were several constraints in our study, most of which are common to retrospective analysis of diagnostic cases. An obvious pitfall is the current method for diagnosis of ABLD; at present, a diagnosis is achieved by demonstration of periportal necrosis, thus excluding any other potential histologic presentations of the disease. Unfortunately, until an etiologic agent is confirmed, studies of the pathology of ABLD will be limited by this narrow histologic definition. Additionally, the use of diagnostic submissions will bias results toward more severe manifestations of disease. Despite these limitations, ABLD toxin appears to be one of a handful of hepatotoxins that characteristically causes acute, severe, often massive hepatocellular necrosis. Other examples in this region of Australia are limited to cyanobacteria, 2 copper poisoning, usually secondary to parenteral administration, 10 and, although rarely implicated in hepatotoxicity in livestock, amatoxins produced by Amanita sp., Galerina sp., and Lepiota sp. of fungi. 21
ABLD is characterized by striking acute periportal hepatic necrosis, which may extend to massive necrosis in a large proportion of cases, distinguishing ABLD from other well-characterized hepatotoxicoses in ruminants. These features result in an acute clinical syndrome characterized by nonspecific signs of acute hepatic disease, namely a combination of sudden death, photosensitization, recumbency, progressive neurologic signs (hepatic encephalopathy), hypogalactia, and jaundice. Microscopically, hepatotoxins produce a limited variety of lesions within the liver. Centrilobular (periacinar, zone 3) hepatocytes are particularly susceptible to toxin-induced injury for a number of reasons; they are the farthest from the arterial blood supply, contain lower concentrations of reduced glutathione, and are rich in microsomal enzymes, such as cytochrome-P450 enzymes. 14 Zone 3 hepatocytes are therefore at higher risk of hypoxic injury, oxidative injury, and damage caused by toxic metabolites. 14 In contrast, the periportal zone (zone 1) is rich in periportal-specific enzymes, reduced glutathione, and oxygen, and is the primary site of gluconeogenesis. 14 These features result in greater regenerative capacity and reduced susceptibility to hypoxic injury, but also higher levels of oxygen for oxygen-dependent bioactivation and formation of reactive oxygen species (ROS). 14 It has been suggested that periportal necrosis may result in more severe disease because of the role of zone 1 in regeneration and the potential for ROS formation. 6
Toxins capable of causing periportal necrosis are more commonly encountered in human medicine, and include 2-chloropropane, albitocin, alloxan, allyl compounds, cocaine, manganese compounds, pemphigus vulgaris toxin, ferrous sulfate, lamotrigine, mesalamine, phosphorus, synthalin, and acetic acid.15,19 Of the few compounds with known toxic mechanisms, the majority do not require bioactivation, and cause periportal injury via the direct cytopathic effect of the highest concentration of toxin carried by the portal blood.7,18
We observed variable evidence of vascular endothelial injury in a number of cases. Endothelial loss and attenuation of central veins was attributed to agonal hypoxic injury; supported by reversible cell injury in perivenular hepatocytes. Spillover of toxic molecules or bioactive metabolites from damaged hepatocytes is likely to have contributed to the changes affecting portal veins; however, primary endothelial damage by ABLD toxin cannot be excluded.
Lesions in other organs were uncommon; however, the presence of mild-to-moderate renal tubular epithelial degeneration and necrosis in 4 of 14 cases was a notable feature. Given the patchy and varied distribution of lesions in these cases, and the involvement of both proximal and distal convoluted tubular epithelium, it seems more likely that these findings are secondary to hypoxic or ischemic injury during the agonal stages of disease, rather than by the action of ABLD toxin. Although involvement of the kidneys could not be excluded in our cases, renal involvement does not appear to be of primary importance in the pathogenesis of ABLD.
Clinical signs of ABLD were consistent with acute hepatic failure. The rapid development and progression of ABLD is unusual among hepatotoxicants, helping to differentiate it clinically from other regionally more common hepatotoxic diseases, such as sporidesmin toxicosis (facial eczema, pithomycotoxicosis), which is a primary differential for secondary photosensitization in ABLD-affected regions. 1 Mid-lactation dairy cows were over-represented in our case series, consistent with previous reports of risk factors for ABLD 16 ; lactating dairy cattle in southern Australia tend to be rotationally grazed, a factor that may increase the risk of toxin exposure.
A key finding was the magnitude of hepatocellular enzyme elevations compared to cholestatic enzyme elevations. GLDH, a sensitive and specific marker for hepatocellular injury in ruminants, 12 was frequently increased by > 20 times the upper RI in cases of ABLD; cholestatic enzyme activities such as GGT, although markedly elevated, were significantly lower than GLDH in all cases. These enzyme derangements are not unique to ABLD, but in cases in which histopathology is not available, may be useful for differentiation of ABLD and pithomycotoxicosis, which does not typically cause such marked elevations of GLDH activity. 4 All other hematologic and biochemical changes were nonspecific and attributable to disease severity (stress, dehydration, inappetence) and/or the metabolic requirements of lactation.
Evidence of hemorrhagic diathesis was the most common macroscopic finding. Coagulopathies are frequent sequalae to acute hepatotoxicity, but the exact mechanism is unclear. 11 In cases of acute hepatotoxicity, it is suspected that overwhelming hepatic injury results in uncontrolled consumption of clotting factors and subsequent disseminated coagulopathy. 3 Diminished synthesis of clotting factors with short half-lives (factors V, VII, IX, X), diminished clearance of coagulation by-products, and abnormal fibrinogen synthesis and thrombocytopathy as a result of metabolic disturbances and reduced clearance of fibrin and/or fibrinogen degradation products by the diseased liver may also contribute. 3 In our study, a number of cases had thrombocytopenia and hepatic thrombosis; supporting the possibility of uncontrolled consumption of clotting factors within the diseased liver.
Case 44 provided an insight into the interaction between pithomycotoxicosis and ABLD, with evidence of ABLD in the majority of the liver, and sporidesmin targeting the left liver lobe. The mechanism for preferential damage to the left liver lobe in cases of pithomycotoxicosis is not well understood but is thought to be the result of portal streaming. 3 This case demonstrates that, counterintuitively, chronic diseases such as pithomycotoxicosis may have a protective effect against the toxicant, possibly because of reduced toxin agent delivery to zone 1 hepatocytes.
We have provided here an initial insight into the chronology of ABLD lesions. Long-term survivors are not readily available for study, likely because of minimal incentive for follow-up in recovered cases. Cases 24 and 25, the only individuals to survive an initial outbreak, had primarily cytoplasmic vacuolation affecting the periportal and midzone, with only small foci of periportal necrosis. This may reflect a lower level of initial toxin ingestion, allowing prolonged survival. Notably, fibrosis was absent in both cases, supporting the assumption that ABLD occurs as a result of rapid intoxication followed by death or recovery, rather than an acute-on-chronic or chronic process. Both cases also had cellular changes that were not present in acute cases, namely anisocytosis, anisokaryosis, and rare megalocytosis with occasional binucleation. The hepatocellular replicative response may partly explain the cellular pleomorphism seen in these cases. This does not explain the rare megalocytosis and multinucleation, which may be the result of the longer-term effects of ABLD toxin, or may be confounded by the effects of other chronically ingested hepatotoxic agents, such as pyrrolizidine alkaloids or aflatoxin B1, which cannot be excluded.5,17 More work is required to determine not only the etiologic agent, but also the full spectrum of lesions associated with disease.
Supplemental Material
sj-pdf-1-vdi-10.1177_10406387211025829 – Supplemental material for Clinical and pathologic features of acute bovine liver disease in Australia
Supplemental material, sj-pdf-1-vdi-10.1177_10406387211025829 for Clinical and pathologic features of acute bovine liver disease in Australia by Eve M. Manthorpe, Ian V. Jerrett, Grant T. Rawlin and Lucy Woolford in Journal of Veterinary Diagnostic Investigation
Footnotes
Acknowledgements
We thank Dr. John Gibney for his clinical and epidemiologic insights, and Dr. Kandarp Patel for his technical assistance with the figures.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.