
Abstract
The value of next-generation sequencing (NGS)-based applications for testing purposes in human medicine is widely recognized. Although NGS-based metagenomic screening may be of interest in veterinary medicine, in particular for intensively farmed livestock species such as pigs, there is a lack of protocols tailored to veterinary requirements, likely because of the high diversity of species and samples. Therefore, we developed an NGS-based protocol for use in veterinary virology and present here different applications in porcine medicine. To develop the protocol, each step of sample preparation was optimized using porcine samples spiked with various RNA and DNA viruses. The resulting protocol was tested with clinical samples previously confirmed to be positive for specific viruses by a diagnostic laboratory. Additionally, we validated the protocol in an NGS viral metagenomics ring trial and tested the protocol on viral multiplex reference material (NIBSC, U.K.). We applied our ViroScreen protocol successfully for 1) virus identification, 2) virus characterization, and 3) herd screening. We identified torque teno sus virus and atypical porcine pestivirus in a neurologic case, determined the full-length genome sequence of swine influenza A virus in field samples, and screened pigs using pen floor fecal samples and chewing rope liquid.
Keywords
Introduction
Many different tools such as sequence-specific PCR and ELISAs for antigens or antibodies are available for the detection of viruses in veterinary diagnostic laboratories. However, these assays require careful triage and a targeted approach given that they are designed for and limited to circumscribed groups of known viruses.
Given the high demand for pork meat, the pig production industry is constantly growing. 36 A disease outbreak caused by a new or emerging virus may lead not only to massive economic losses but could also endanger human health; pigs were shown to be an important source of zoonotic viruses such as Nipah virus in Malaysia 39 or the influenza A (H1N1) virus (IAV) that caused the “swine flu” pandemic.14,51 Therefore, having an efficient approach available that allows detection of known viruses but also of novel or unexpected viruses in a single test could be crucial to recognize and manage viral outbreaks in pigs worldwide.
Next-generation sequencing (NGS) technologies, particularly suitable for metagenomic analyses, opened a new chapter in viral detection and identification. Using NGS, detection of nucleic acids of novel and known viruses is possible without specific targeting or knowledge of the genome. Moreover, the whole range of viruses present in a sample can be identified simultaneously, reducing the number of tests that must be performed. As an additional benefit, NGS may provide fast full-length virus genome sequencing. Therefore, NGS is an ideal technology to use for submissions in which routine tests fail to identify a viral etiology, but also for research studies that aim to characterize the viral community in a specific environment, the so-called virome. For example, NGS has enabled detection of Schmallenberg virus in cattle, 23 a torque teno sus virus (TTSuV) and a novel porcine parvovirus in pigs with postweaning multisystemic wasting syndrome, 7 and a novel astrovirus in a mink with shaking mink syndrome. 8
Although NGS is already used routinely in the human medical field for applications such as detection of cancer markers in oncology 26 or identification and characterization of pathogens in infectiology, 16 in the veterinary field, NGS is still reserved primarily for research-related applications. One point of hindrance may be the lack of reliable and approved methods applicable to a broad range of sample types and species, from sample preparation to data analysis. For NGS-based virus detection in humans, several sample-preparation protocols have been published.12,30,34 However, these protocols often focus on blood or cerebrospinal fluid. In contrast, NGS analysis in veterinary cases is often done postmortem, and sample materials range from various solid tissues to blood and all secretions and excretions. 13
We have developed and validated a protocol termed “ViroScreen” for sample preparation and metagenomic analysis for the detection of viruses in veterinary samples. Given that sequencing of total nucleic acid of cell-rich material results primarily in host genomic sequences, with the viral sequences making up only a minuscule percentage of the reads, we tested the validity of a relative enrichment for viral particles using spiked-in sample material. Next, sample material known to be positive for specific viruses was sequenced and compared to amplicon sequencing. For validation of the method, a commercial highly multiplexed viral pathogen reagent containing 25 different viruses was sequenced, and we participated in a ring trial for viral metagenomic analyses. Finally, we evaluated the applicability of the method for pig herd screening by comparing sequencing results from pen floor feces from a group of animals to single animal sampling.
Material and methods
Animals and samples
Swine clinical samples such as feces, lung tissue, and nasal swabs were obtained from the Institute of Veterinary Pathology, Vetsuisse Faculty, University of Zurich (VSF-UZH). Samples from naturally infected animals and swine IAV-positive nasal swabs were provided by the Diagnostic Department of the Institute of Virology, VSF-UZH. For validation, a commercial viral multiplex reagent (VMR 11/242-001; National Institute for Biological Standards and Control [NIBSC], South Mimms, U.K.) containing 25 human viruses was tested. For the herd screening trial, pen floor feces and individual samples from pigs kept in this pen (i.e., feces from rectum, mixed feces from the floor, chewing rope fluid, nasal swabs, bronchoalveolar lavage (BAL) fluid, and blood samples) were collected. Pigs were anesthetized with an intramuscular injection of ketamine (Narketan, 10 mg/kg; Vetoqinol) and azaperone (Stresnil, 2 mg/kg; Elanco). Nasal swabs were collected by flocked swabs (FLOQSwabs 519CS01; Copan) and submerged in 1 mL of phosphate-buffered saline (PBS). BAL was performed in anesthetized animals via laryngoscope (Heine) after introducing a catheter (SMS medipool) through the mouth into the trachea. Fifteen mL of sterile sodium chloride solution was injected using a 20-mL disposable syringe and aspirated immediately. Additionally, blood was collected from the anesthetized pigs from the jugular vein into vacutainers (serum, 7.5 mL, S-Monovette; Sarstedt), and feces was sampled from the rectum with a small spoon into a stool tube (15 mL; Boettger). All samples were collected for veterinary student training purposes by the Division of Swine Medicine, VSF-UZH, and were frozen promptly after collection and stored at −20°C until used further.
Viruses and specific detection assays
For the spiking experiments, 6 cell culture–grown viruses from different families and with different types of genomes and particle sizes were used: bovine viral diarrhea virus (BVDV; Pestivirus), bovine herpesvirus 1 (BoHV-1; Bovine alphaherpesvirus 1), swine IAV, porcine rotavirus A (PRV-A), porcine parvovirus (PPV; Ungulate protoparvovirus 1), and transmissible gastroenteritis virus (TGEV; Alphacoronavirus 1; Table 1). BVDV was isolated from leukocytes of the persistently infected animal “Carlito” 47 and passaged 6 times on MDBK (Madin–Darby bovine kidney) cells. BoHV-1 was isolated from the last outbreak of infectious bovine rhinotracheitis in Switzerland 6 and passaged 7 times on MDBK cells. The swine IAV strain PR-8 (A/Puerto Rico/8/1934(H1N1)) was kindly provided by Jovan Pavlovic (Institute of Medical Virology, University of Zurich) and passaged on Madin–Darby canine kidney cells. The PRV-A strain OSU was kindly provided by Catherine Eichwald (Institute of Virology, VSF-UZH) and passaged on MA104 cells (embryonic rhesus monkey kidney). 18 PPV had been isolated from a diagnostic positive sample at the Institute of Virology, VSF-UZH, and passaged 6 times on porcine kidney cells (PK13). The TGEV strain Purdue was originally provided by Maurice B. Pensaert (Laboratory of Virology, Faculty of Veterinary Medicine, Ghent University, Belgium) in the 1980s, passaged countless times on PK13 cells, and used as positive control for TGEV reverse-transcription real-time PCR (RT-rtPCR) in the Diagnostic Department of the Institute of Virology, VSF-UZH. The cycle quantification (Cq) values in the supernatant after spiking were as follows: BoHV-1: 27; BVDV: 26; swine IAV: 27.5; PPV: 21; PRV-A: 16; and TGEV: 23.8. The aim was to detect all of the viruses regardless of the Cq value.
Main features of viruses used for spiking experiments and present in samples from natural infections.
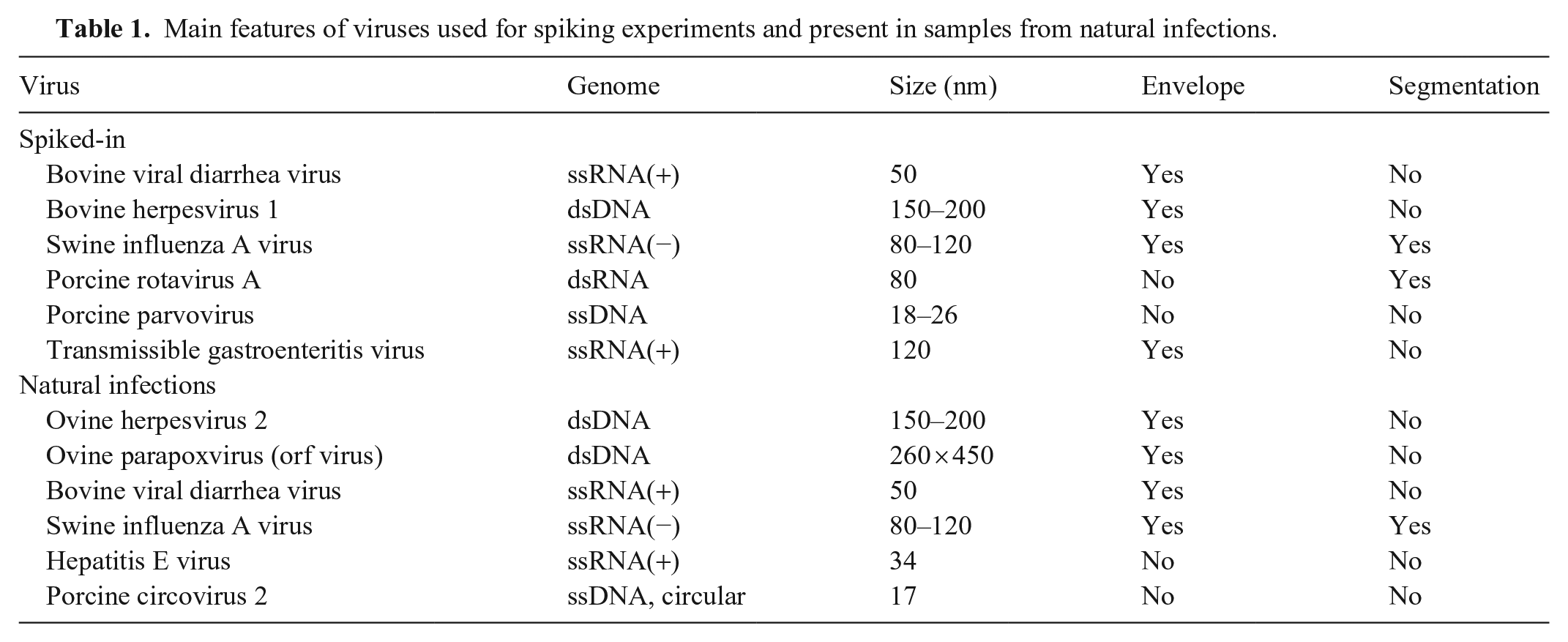
Considering that spiked-in viruses do not fully reflect a natural infection in which virus is cell-associated or even intracellular, we decided to also sequence clinical samples that had tested positive previously for specific viruses by commercial and in-house rtPCRs at our Diagnostic Department, VSF-UZH (Table 1). Swine IAV was tested by a commercial test (Adiavet SIV RT-PCR kit; Bio-X Diagnostics). The following 4 viruses were tested with published PCRs: ovine herpesvirus 2, 24 parapoxvirus, 40 hepatitis E virus (HEV), 33 and BVDV. 49 The porcine circovirus 2 (PCV-2)–positive liver had been confirmed by immunohistochemistry at the Institute of Veterinary Pathology, VSF-UZH.
Sample preparation
The general sample preparation method for ViroScreen is described here. Adaptations and samples used for specific experiments are described in detail further below.
Solid samples were chopped into small fragments with a scalpel blade, weighed, and diluted 1:10 in PBS. For liquid samples, 300 µL were directly used in the second step, which consisted of homogenization (TissueLyser II; Qiagen) for 2 min at 20 Hz. Solid samples, such as skin crusts, were additionally homogenized with a stainless-steel bead (5 mm; Qiagen). After homogenization, samples were centrifuged at 16,060 × g for 5 min, and the resulting supernatants were passed through a 0.45-µm syringe filter to remove host cells and most bacteria (Puradisc, 13 mm; Whatman GE Healthcare). Then, 134 µL of the filtered supernatant was treated with 1 µL of micrococcal nuclease (New England Biolabs) and 1 µL of ribonuclease A from bovine pancreas (Merck) in 14 µL of micrococcal nuclease buffer (New England Biolabs) for 15 min at 45°C followed by 1 h at 37°C to digest free-host DNA and RNA, respectively. Total nucleic acid was isolated (QIAamp viral RNA mini kit; Qiagen) according to the manufacturer’s manual, except that the carrier RNA was omitted and β-mercaptoethanol (Bio-Rad) at a final concentration of 1% was added to the AVL buffer (Qiagen) to inactivate the nucleases.
To sequence RNA and DNA simultaneously, the RNA in the samples was reverse-transcribed into cDNA using 2.5 µmol of random primer with a known 20-nt tag sequence at the 5′-end (SISPA-N: GTTGGAGCTCTGCAGTCATCNNNNNN) and the RevertAid First Strand H minus cDNA synthesis kit (Thermo Fisher) according to the manufacturer’s manual. Thereafter, 1 µL of RNase H (New England Biolabs) was added to degrade residual RNA. A premix of 0.8 µM SISPA-N primer, 10× Klenow buffer, and 0.2 mM dNTP was added to 45.5 µL of the first-strand DNA. After denaturing at 95°C for 1 min followed by cooling on ice, the second strand was synthesized with 5 U/20 µL of Klenow polymerase (Thermo Fisher) for 15 min at 25°C followed by 1 h at 37°C. An additional step of second-strand synthesis using Klenow polymerase was performed under the same conditions, followed by DNA purification (PureLink PCR micro kit; Invitrogen) and elution in 12 µL of elution buffer. To amplify the double-stranded DNA nonspecifically, a sequence-independent single primer amplification (SISPA) was performed. For this, the HotStarTaq DNA polymerase (Qiagen) and the SISPA primer (GTTGGAGCTCTGCAGTCATC) that binds to the reverse-complement sequence of the tag introduced with the SISPA-N primer were used under the following conditions: 15 min of activation at 95°C, 18 cycles of 30 s at 94°C, 30 s at 58°C, and 1 min at 72°C, followed by 10 min at 72°C and cooling to 4°C. The amplified products were purified (QIAquick PCR purification kit; Qiagen), eluted in 30 µL of EB buffer (Qiagen), and the total DNA concentration was measured (Qubit fluorometer; Invitrogen) using the DNA high-sensitivity assay.
Library preparation and sequencing
Prior to sequencing, the DNA was fragmented (E220 Focused-ultrasonicator; Covaris) to ~ 500-bp fragments. Sequencing libraries were constructed (NEBNext Ultra I kit, or the NEBNext Ultra II kit; New England Biolabs). For barcoding, the NEBNext Multiplex Oligos (New England Biolabs) were used. All samples were sequenced (NextSeq500 sequencer; Illumina) at the Functional Genomics Center Zürich (FGCZ) in 2 × 150-bp paired-end run using the medium-output flowcell for 14–16 samples or high-output flowcell for 28 samples in one run, to obtain at least 5 million sequencing reads per sample.
After quality control (FastQCapp; SUSHI, FGCZ) 20 and de-barcoding, the reads were assembled to the latest available NCBI virus reference sequence database (ftp://ftp.ncbi.nlm.nih.gov/refseq/release/viral/) and to a self-designed extensive database that contained 61,620 complete viral genome sequences with a genome size range of 500 bp to 300 kbp downloaded from the NCBI nucleotide database. Known plant viruses and bacteriophages were omitted from our database. To test more specifically (e.g. to control for spiked-in viruses), purposefully assembled sequence databases containing only the searched viral strains were used. To align the reads to the databases, the metagenomic pipeline of the SeqMan NGen software v.14–v.16 (Lasergene; DNAStar) was used. This software and pipeline have recently been used for viral metagenomic analysis from farmed mink 50 and for studying the evolution of Ebola virus. 1 Assembled sequences were visualized and analyzed (SeqMan Pro software, Lasergene; DNAStar). As positive control for the whole protocol as well as for control of contamination, the same lung tissue known to be weak positive for TTSuV was prepared and sequenced along with the other samples in all runs.
Spiked samples
To establish ViroScreen as a veterinary viral detection tool using NGS, several crucial steps of sample preparation needed to be addressed. Given the large amount of non-viral nucleic acids, it was likely that samples would profit from relative enrichment for viral particles to increase the sensitivity of virus detection. Therefore, samples spiked with a range of known viruses were prepared with and without enrichment, and the recovery of viral sequences was compared. The samples consisted of porcine nasal swabs, feces, and lung tissue, and were spiked with 6 known cell culture–grown DNA and RNA viruses of different genome sizes (Table 1). The samples were divided into 4 groups, according to the sample preparation. The first group (GR1) was prepared with relative enrichment for viral particles (filtration and nuclease treatment) and random DNA amplification (SISPA); the second group (GR2) was prepared with enrichment for viral particles but without subsequent random amplification; the third group (GR3) was amplified without prior viral enrichment; and in the fourth group (GR4), enrichment for viral particles and amplification were omitted altogether.
Sequencing of clinical samples
Spleen of a bison positive for ovine herpesvirus 2 (OvHV-2), skin crust of a sheep positive for ovine parapoxvirus (orf virus), spleen of a cow persistently infected with BVDV, pig liver strongly positive for PCV-2 by immunohistochemistry (IHC), a porcine nasal swab positive for swine IAV, and finally a human stool sample and a raw pork sausage positive for HEV by RT-rtPCR were selected and prepared according to our ViroScreen protocol. Additionally, a brain sample of a 4-mo-old piglet with neurologic signs, in which histology revealed nonsuppurative encephalitis, suggesting a viral cause, was also prepared.
Comparison of targeted and non-targeted whole-genome sequencing
All 8 swine IAV genome segments were specifically amplified from a positive nasal swab (Cq of 25; PathAmp FluA RT-PCR kit; Thermo Fisher). The same nasal swab was prepared nonspecifically according to our ViroScreen protocol. Both DNA samples were subjected to library preparation and sequenced as described above on an Illumina NextSeq machine in the same run. The resulting reads were assembled to the NCBI RefSeq database as well as a swine IAV–only database. The number of aligned reads and the genome coverage of the best matching reference was compared between the PCR-amplified sample and the sample prepared nonspecifically (ViroScreen).
Validation with external samples
We used our ViroScreen protocol to prepare 5 serum samples spiked with an unknown number of viruses in the context of the first Swiss-wide viral metagenomic interlaboratory proficiency test, 25 and to sequence a VMR containing 25 viruses using 150 µL of the VMR as input material for sample preparation. The sequencing was performed in triplicate and analyzed in 2 ways: 1) including all reads available, and 2) using only a subset of 2 million reads. This allowed us to compare our results to those from a published interlaboratory test performed with the same VMR. 37
Pig pen screening
We also tested our method for virus screening of a whole group of animals. We were particularly interested in the suitability of chewing rope fluid and feces from the pen floor to represent the virome of the whole group and hence wanted to compare the virus findings in these pen floor samples to the viruses present in samples taken directly from the animals. From each animal in a bay of seven 7-wk-old piglets, a blood sample, nasal swab, rectal fecal sample, and BAL sample were taken. Feces was collected from the pen floor, and a cotton chewing rope was placed in the bay for 30 min for the piglets to play with and chew. Samples from 3 and 4 individual animals were pooled prior to sample preparation. For example, 30 mg of individual feces from animals 1–3 were mixed to form feces-pool 1, and 30 mg of animals 4–7 formed feces-pool 2. For the liquid samples, 100 µL were used for pooling. The chewing rope liquid was manually squeezed from the rope into a plastic bag and transferred into a tube. All pools and the pen floor sample were prepared according to our ViroScreen protocol: fecal samples were diluted 1:10 in PBS; liquid samples were used undiluted. Hence, a total of 8 libraries from the individual samples and 1 library each from the pen floor and chewing rope were prepared.
Results
Testing the efficiency of virus enrichment
The data obtained from sequencing 3 types of sample material (i.e., feces, nasal swab, and lung tissue), spiked with 6 viruses, showed that the relative number of viral reads, expressed in percentage of total reads, was highest in samples enriched for virus particles and subsequently amplified (GR1; Fig. 1). The spiked-in viruses represented 67% of the total number of reads in the nasal swab, 27% in lung tissue, and 3% in feces, when mapped to the spiked-in viruses. In GR2, the relative number of the viral reads in nasal swab and lung was higher than in the GR3, in which only amplification was applied. In all types of material, the relative number of reads recovered from samples without any pretreatment was the lowest, with values < 0.5%. However, the effect of enrichment differed between the different spiked-in viruses (Suppl. Fig. 1). Although the combination of enrichment and amplification clearly enhanced the detection of BVDV, PRV-A, swine IAV, PPV, and TGEV, this was not the case for BoHV-1. For BoHV-1, enrichment alone without subsequent amplification was more effective than the combination of enrichment and amplification, except for BoHV-1 spiked into feces, in which the untreated sample had the highest number of relative reads.
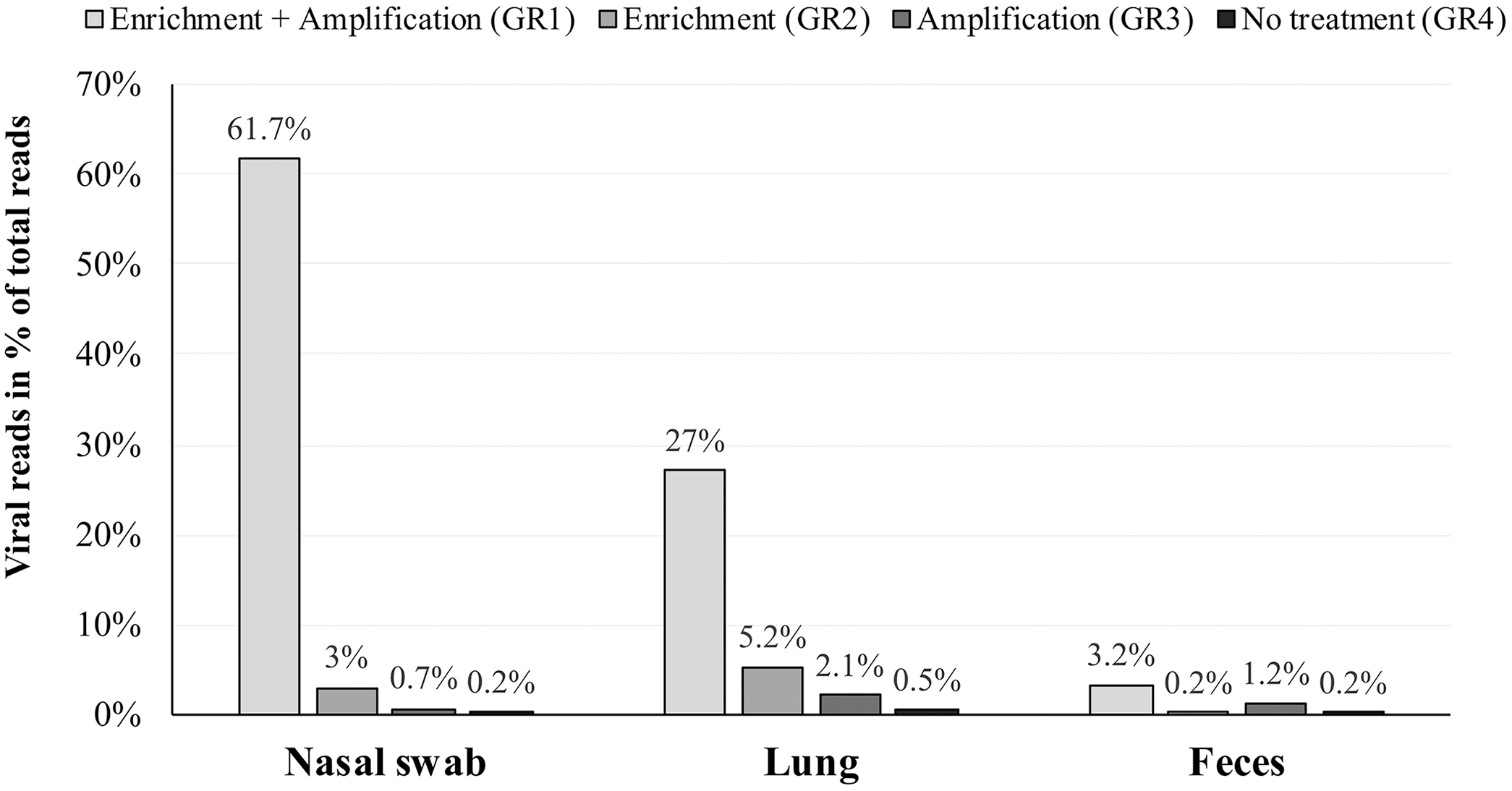
Percentage of viral reads in relation to total reads in next-generation sequencing data of nasal swab, lung tissue, and feces samples spiked-in with 6 viruses. Samples were prepared in 4 different combinations, with or without enrichment for viral particles and amplification. GR = group.
Sequencing of clinical samples
Our ViroScreen protocol confirmed the presence of viruses detected previously by specific PCRs or by IHC (Table 2). In the spleen sample and sheep skin crust, 73 OvHV-2–positive and 1,540 ovine parapoxvirus–positive reads were detected, respectively. From the spleen of a persistently infected cow, 10,501 reads of BVDV were assembled. In the swine IAV–positive nasal swab, all 8 viral genome segments were sequenced with 100% genome coverage, and the number of aligned reads ranged from 195 for the 7th segment (proteins M1 and M2) to 3,835 in the 1st segment (polymerase-based protein 1 [PB1]). In the human stool sample and raw pork sausage, the entire genome of a Swiss-specific subtype of HEV-3 has been confirmed. 5 The sequences from these 2 epidemiologically related HEV-3–positive samples were identical, except for a few single-nucleotide variations (SNVs). In the PCV-2 antigen–positive liver tissue, 10 million reads were aligned to the PCV-2 reference sequence (Table 2). Finally, in the brain from a pig with nonspecific neurologic signs, 15,394 reads of TTSuV-1 and 96.2% genome coverage, and 2.5 million reads and a fully covered genome of atypical porcine pestivirus (APPV), were detected. Both viruses were confirmed by specific rtPCRs.28,42
Read counts and Cq values of samples from natural infections tested by specific real-time PCR (rtPCR) and ViroScreen metagenomic sequencing.
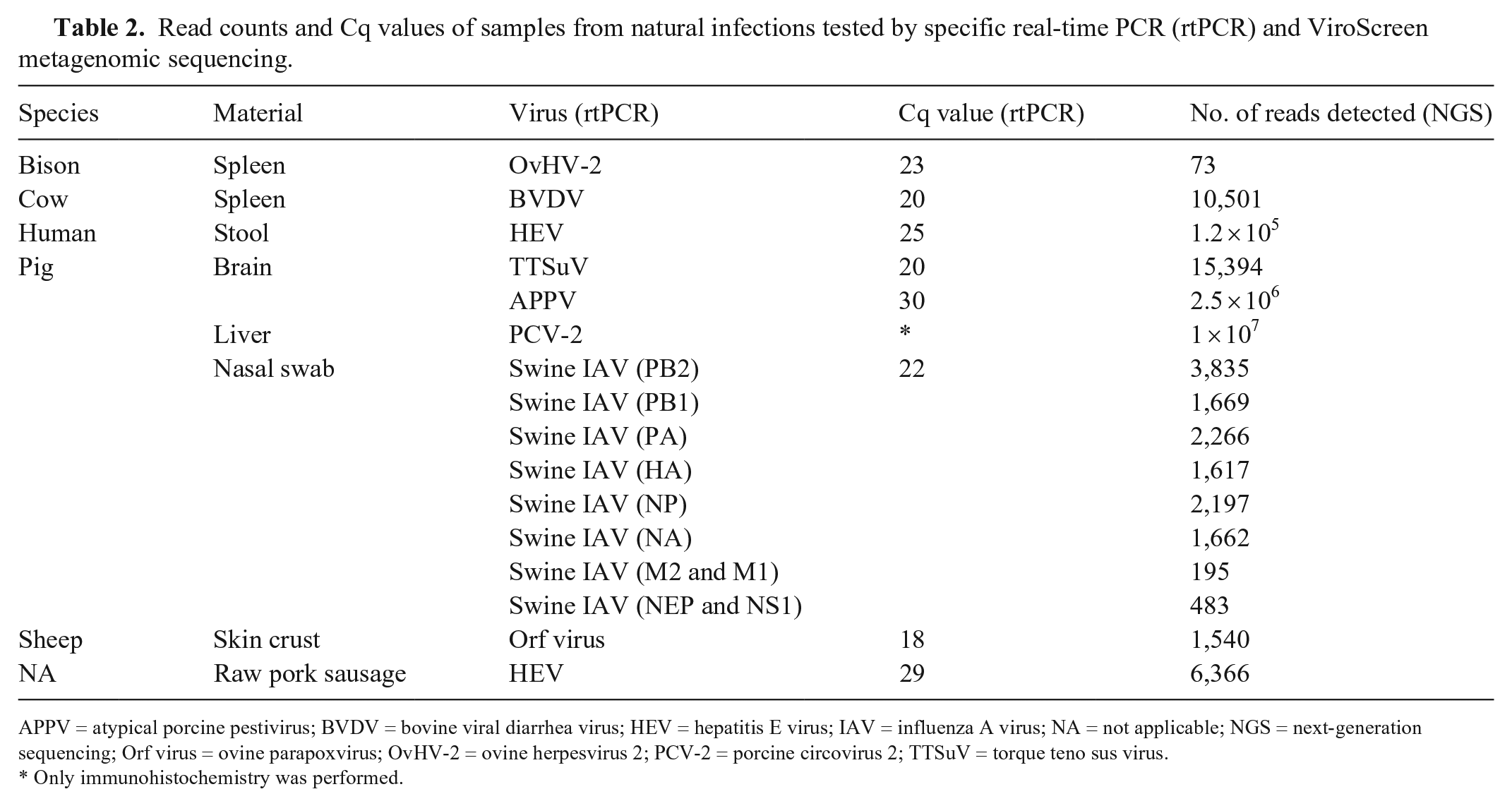
APPV = atypical porcine pestivirus; BVDV = bovine viral diarrhea virus; HEV = hepatitis E virus; IAV = influenza A virus; NA = not applicable; NGS = next-generation sequencing; Orf virus = ovine parapoxvirus; OvHV-2 = ovine herpesvirus 2; PCV-2 = porcine circovirus 2; TTSuV = torque teno sus virus.
Only immunohistochemistry was performed.
Comparison of targeted and non-targeted whole-genome sequencing
Not surprisingly, more reads (1.16 million) were assembled to the reference sequence in the sample prepared using the PathAmp FluA RT-rtPCR than in that prepared by the ViroScreen protocol (0.83 million), particularly for the shorter segments (Fig. 2). For 3 longer segments, more reads were aligned from our ViroScreen protocol than from targeted sequencing. Both methods enabled whole-genome sequencing (WGS) given that all 8 swine IAV segments were fully covered. The sequences were almost identical, except that ViroScreen revealed more SNVs, which may represent quasispecies variability and may support evolutionary studies and mixed swine IAV infection analyses. Subsequent sequencing of more swine IAV–positive nasal swabs has shown that full-genome sequencing by ViroScreen is possible up to Cq values of 26–28; at higher Cq values, the coverage, especially of shorter segments, drops (data not shown).
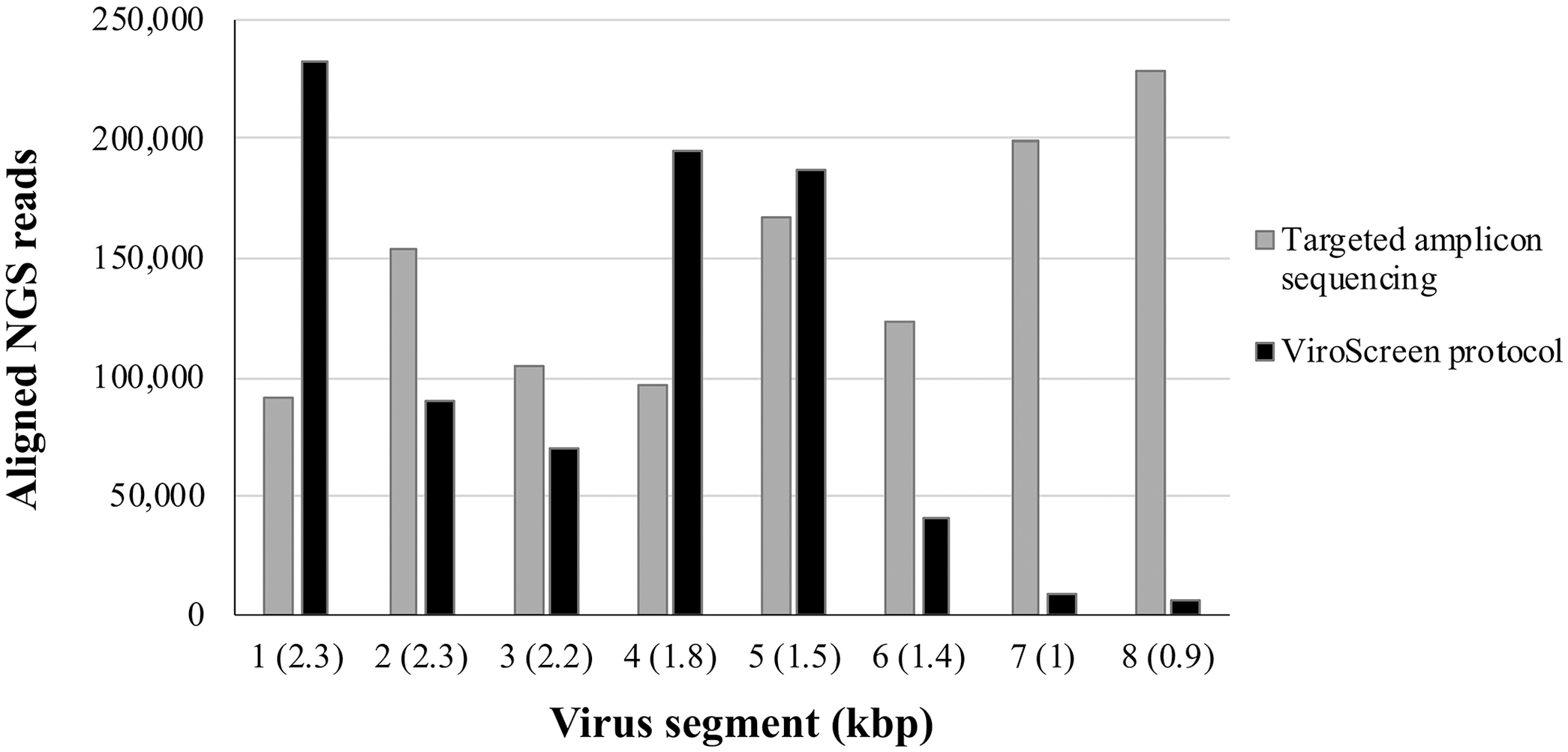
Number of reads aligned to each segment of swine influenza A virus from a porcine nasal swab prepared by targeted amplicon sequencing method and ViroScreen protocol. Virus segments: 1 = polymerase (PB2); 2 = polymerase (PB1); 3 = polymerase (PA); 4 = hemagglutinin (HA); 5 = nucleocapsid protein (NP); 6 = neuraminidase (NA); 7 = matrix protein 2 (M2) and matrix protein 1 (M1); 8 = nuclear export protein (NEP) and nonstructural protein 1 (NS1).
Validation with external samples
The VMR containing 25 viruses was sequenced in triplicate resulting in 14, 17, and 14 million reads in each replicate. When analyzing all reads available, 24, 23, and 23 of 25 viruses were detected, respectively (Table 3, Suppl. Table 1). Norovirus genogroup I (GI) was not detected in any replicate. Additionally, influenza B virus and norovirus GII were not detected in the second and third replicate, respectively. The maximum number of detected viruses when using only 2 million reads was 22, 21, and 21 for each replicate. Norovirus GI, GII, and sapovirus were not detected in any replicate. In addition, influenza B virus and human coronavirus were not detected in the second and third replicate, respectively.
Number of viruses (of 25) detected in the multiplexed viral pathogen reagent (Suppl. Table 1) from all obtained reads and a subset of 2 × 106 reads, and identity of missed viruses.

HCoV = human coronavirus; IBV = influenza B virus; NoV GI, NoV GII = norovirus genogroup I and II, respectively; SoV = sapovirus.
Given that the detailed results of the Swiss interlaboratory proficiency test for viral metagenomics organized by the Swiss Institute of Bioinformatics trial have been published, we only briefly summarize here our performance. Three of the 5 samples were serum of a healthy human donor spiked with a mix of 4 viruses, including human betaherpesvirus 5, human mastadenovirus B, enterovirus C, and IAV in increasing dilutions (1:1, 1:10, 1:100). The fourth sample was a commercial multiplex reference reagent containing 11 human viruses related to immunodeficiencies, herpesviruses, polyomaviruses, an adenovirus, and a parvovirus strain (15/130-XXX; NIBSC). The fifth sample was a negative control reagent consisting of a pool of human sera that tested negative for specific viruses such as HIV, and hepatitis B and C viruses (11/B606; NIBSC). We detected all 4 viruses spiked into serum in all dilutions. However, in 2 samples, we had some false-positive matches, including human papillomavirus and parvovirus NIH-CQV. These also showed up in the virus-negative sample, along with low read numbers of poliovirus, human adenovirus, and betaherpesvirus 5, representing contamination by viruses present with high read numbers in the other samples. From the 11 viruses in the multiplex control, only 4 were correctly detected. All 7 missed viruses were human herpesviruses with Cq values ≥ 30, such as herpes simplex virus 1 and 2 (Human alphaherpesvirus 1 and 2), Epstein-Barr virus (Human gammaherpesvirus 4), human cytomegalovirus, and varicella zoster virus (Human alphaherpesvirus 3).
Pig pen screening
Sequencing feces, nasal swabs, lung lavage, and serum collected from individual animals revealed 18 distinct viruses of porcine origin (Table 4). Most of these (12 viruses) were detected in fecal samples and serum (6 viruses). The spectrum of viruses present in fecal samples is clearly different from the spectrum discovered in nasal swabs and BAL. The most frequent viruses in fecal samples were porcine serum–associated circular viruses, porcine bocaparvovirus, and porcine astrovirus (Mamastrovirus 3). Although many enteric viruses were sequenced in fecal samples, nasal swabs seem to be the “cleanest” type of material concerning viral reads, given that only 3 viruses were detected with a low number of reads and coverage (i.e., porcine bocaparvovirus 2 and 5, and circular Rep-encoding ssDNA [CRESS] viruses). In serum, only reads assembled to the TTSuV-1 were detected in high numbers. Interestingly, CRESS viruses were only detected in nasal swabs, BAL, and serum, but not in feces. CRESS viruses belong to several different virus families, and their taxonomy is still evolving. 52 Their clinical meaning as well as their biology is, with few exceptions (e.g., porcine circoviruses), largely unknown.
Viruses detected, number of reads assembled, and genome coverage in samples collected from individual animals and 2 naturally pooled samples from a pig pen.
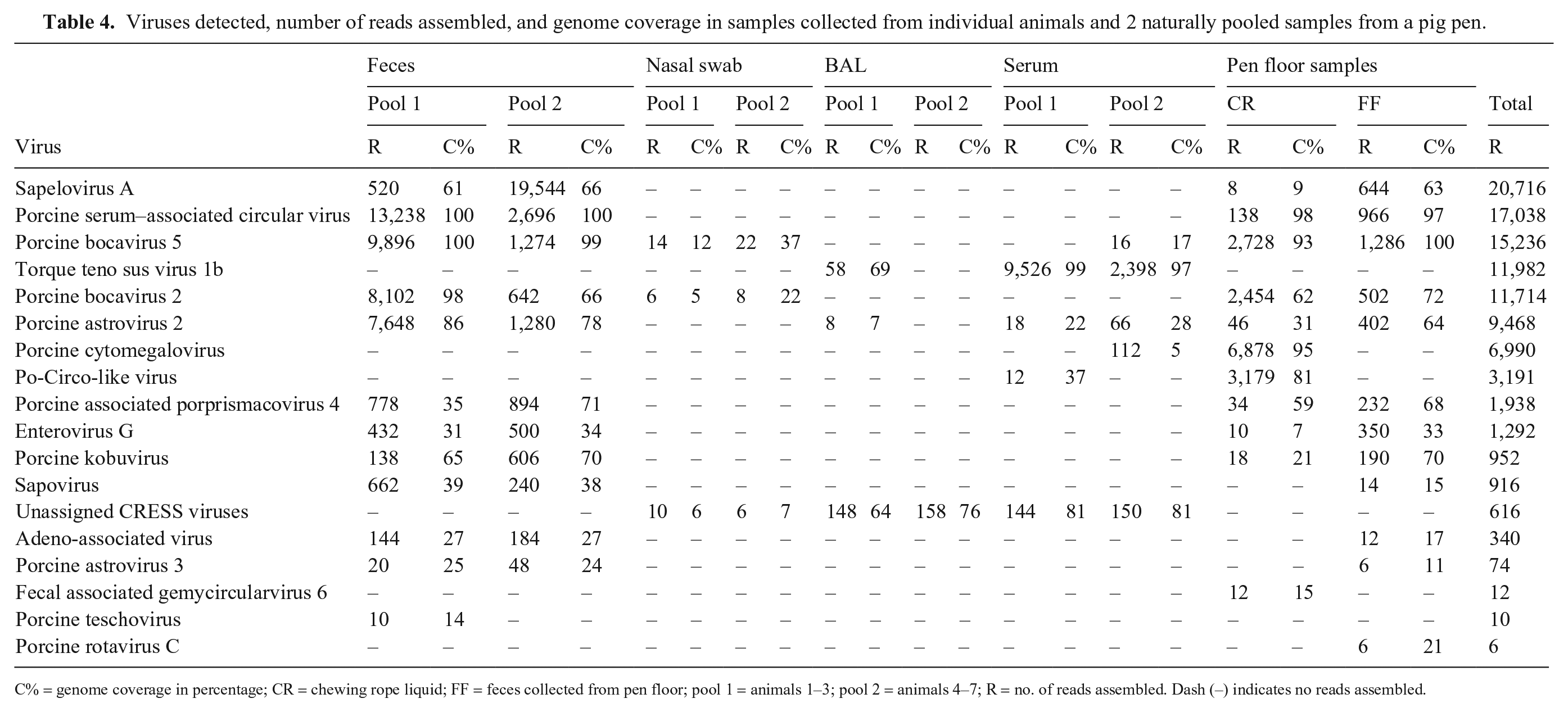
C% = genome coverage in percentage; CR = chewing rope liquid; FF = feces collected from pen floor; pool 1 = animals 1–3; pool 2 = animals 4–7; R = no. of reads assembled. Dash (–) indicates no reads assembled.
We detected 12 viruses in the pen floor fecal samples as well as in the individual fecal samples. Porcine teschovirus was detected only in the pen floor samples, although at low read numbers (10 reads). In contrast, porcine rotavirus C was detected only in the individual samples but also at low read numbers (6 reads). ViroScreen analysis of the chewing rope material yielded 11 different viruses, 8 of these were detected also in pen floor and individual fecal samples. Two of the viruses, porcine cytomegalovirus and porcine circovirus, that were detected in the chewing rope material with 6,878 and 3,179 reads, respectively, were detected also in serum, although at low read numbers (112 and 12, respectively). Fecal-associated gemycircularvirus, also a CRESS virus, was only detected in chewing rope liquid and at low read numbers (12 reads).
Discussion
Our sample preparation protocol for metagenomic virus sequencing presented here is an attempt to apply NGS for routine testing in veterinary diagnostic laboratories. Given that, in metagenomic sequencing, all nucleic acids present in a sample are sequenced, reducing host, bacterial, and other background nucleic acids may significantly enhance the sensitivity of virus detection. Therefore, we first tested the effect of enrichment for viral particles and random amplification. For all types of sample material (nasal swab, feces, and lung tissue), the combination of enrichment for virus particles and random amplification by SISPA resulted in the highest proportion of viral reads. However, although this is true for BVDV, TGEV, PPV, RVA, and swine IAV, the best results for BoHV-1 were achieved with enrichment alone, omitting subsequent amplification. It is not clear why this was the case. Losing the virus by filtration given the relatively large size of the particle cannot be the reason because enrichment worked well with the ovine parapoxvirus, which is even larger. We also excluded a general problem with sequencing DNA viruses, because PCV-2 was well recovered after enrichment and SISPA. It seemed that specifically BoHV-1, or herpesvirus DNA in general, was not well amplified by SISPA. This might be a result of the particularly high GC content of the virus (72.4%) that renders sequencing alphaherpesviruses generally challenging. 9
Given that the recovery of viral reads overall was massively increased by enrichment and subsequent amplification, we decided to implement this approach in our ViroScreen protocol. The effect was most pronounced in nasal swabs, with a 268-fold increase of the relative number of viral reads compared to using no pre-treatment, followed by lung tissue (56-fold) and feces (32-fold), both materials containing a high number of non-viral nucleic acids. In the case of feces, non-viral nucleic acids derive not only from the host but also from bacteria and food components and may contain inhibitors such as humic and tannic acids or bile salts. The detection of viruses in stool samples might be even more challenging than in solid samples. Nevertheless, our protocol has already been used successfully to sequence the entire HEV genome from a human stool sample. 31
After using spiked-in material, we tested our ViroScreen protocol on samples from animals naturally infected with viruses of various sizes, such as the tiny parvo- and circoviruses and the > 20 times larger parapoxviruses, which are among the largest clinically relevant viruses. We not only recovered reads from all sizes of viruses, but also from both RNA and DNA viruses with double-stranded or single-stranded, linear, circular, or segmented genomes, from enveloped and nonenveloped viruses, and from a wide range of naturally infected sample material as variable as skin crust and pork sausage. However, the efficiency of recovery varied between sample material and viruses given that the Cq values were not correlated with the number of the reads detected. When tested in rtPCR, all samples had Cq values of 18–29; the number of reads varied between 119,000 for HEV in a human stool sample (Cq 25) and 73 for OvHV-2 in spleen from a cow suffering from malignant catarrhal fever (MCF; Cq 23). The efficiency of the specific tests may be different, and Cq values cannot be compared directly. In addition, the different samples contain different amounts of contaminating host nucleic acid that could influence the recovery of viral sequences.
The experiment with the spiked-in viruses suggested that the enrichment and amplification-based protocol may not be ideal for sequencing herpesviruses. Furthermore, herpesviruses in their latent form are not encapsidated, and hence virus particle enrichment does not work in these cases. MCF is thought to be caused by an unregulated immune reaction to the latent form of the virus. 38 This may explain why OvHV-2 was not very efficiently recovered by ViroScreen. On the other hand, all human herpesviruses included in the VMR were recovered well. The limitation regarding the discovery of herpesviruses, in particular if latent, has to be kept in mind, and alternative methods, such as specific PCR protocols or pan-herpes PCR, 17 need to be considered for suspected cases. However, when comparing viral reads from an equine lung sample positive for equine herpesvirus 5 with and without enrichment for virus particles, the untreated sample resulted in 74 reads (0.00046% of total reads) and the enriched sample in 30 reads (0.00067%; data not shown). Even though the true viral load was unknown, given that detection was by endpoint PCR only, genome recovery was nearly identical with enrichment or without.
Overall, our sequencing results from the clinical samples were in accordance with the results provided by the specific assays, implying that our ViroScreen protocol may provide a detection tool applicable for various sample materials and species. Interestingly, in the case of a piglet with neurologic signs, 2 viruses not specifically targeted in a routine approach were detected, TTSuV-1 and APPV. The presence of TTSuV-1 has been reported in pig brains. 3 However, the clinical signs may more likely be caused by APPV, which was detected at 2.5 million aligned reads. APPV was discovered by applying NGS, 21 and APPV is known to induce neurologic signs in affected pigs.15,19 Owing to the novelty of this virus, APPV was not included in the primary RT-PCR screening. Subsequent RT-rtPCR confirmed that the sample was indeed positive for both APPV and TTSuV-1.
To analyze genetic diversity and mutations of genomes, targeted amplicon sequencing is an approach widely used for WGS. However, designing and testing of primers may be challenging and time-consuming, especially for longer genomes and highly diverse viruses. Therefore, a non-targeted approach by NGS may be an alternative option. Although non-targeted NGS will not replace targeted sequencing, it may in some cases be very useful (i.e., when the targeted taxonomic unit is too diverse for primer design). Here, we compared non-targeted metagenomic NGS (ViroScreen) and targeted amplicon NGS on a swine IAV–positive nasal swab sample with a Cq of 25. All segments were fully sequenced with both methods, with an average depth of 1,867–29,804. However, shearing the DNA for library preparation can lead to loss of 5′- and 3′-ends and may be the cause of shorter segments being sequenced less deep with non-targeted ViroScreen NGS. In addition, the genome sequence resulting from metagenomic sequencing showed more nucleotide ambiguities. Given that swine IAV is an RNA virus with high quasispecies diversity, and the full-length genome sequence is a consensus of many shorter reads that may represent different viral variants, these ambiguities reflect most likely the intra-host diversity of swine IAV. Hence, free of any selective pressure by specific primers, un-targeted NGS may allow not only the determination of the full-length genome sequence, but also analysis of SNVs and quasispecies diversity. Methods based on SISPA have already been used for full-genome sequencing of bovine parvoviruses, Schmallenberg virus, and human rhinoviruses.2,41,45 As shown previously with avian IAV, the SISPA method resulted in lower depth but better coverage for large segments when compared to targeted sequencing. 11 However, another study found the SISPA method less suitable for full genome sequencing because it introduced an amplification bias and uneven reads distribution over the entire genome. 27
Using the entirety of the sequences for analysis (14–17 million reads) generated by sequencing the VMR containing 25 human RNA and DNA viruses covering various genome sizes and virus families, only norovirus GI was not detected. However, as shown before, 35 the viral load of norovirus GI was so low that, once multiplexed, it was not even detectable by RT-rtPCR (Cq > 40). In another study in which VMR was used for an interlaboratory proficiency test, only 2 of 16 laboratories detected all viruses in at least 1 replicate, and norovirus GI was missed by 75% of the laboratories. 37 The laboratory that was able to identify all viruses (in a subset of 2 million reads) prepared the sample also with nucleases and extracted it in a silica column. However, they used the total volume of the reagents (i.e., 1,000 µL) as input for the sample preparation for each replicate, allowing the generation of 54 million reads, whereas we used only 150 µL. With few exceptions, our 3 replicates identified the same viruses and with similar read counts, showing good reproducibility of our method. Interestingly, all 5 human herpesviruses included in the VMR with Cq values of 28–32 37 were detected readily.
The first Swiss-wide viral metagenomic ring trial pointed out several general issues, such as the impact of the database used and the permanent risk of contamination. 25 Regarding our own performance, we easily detected 3 increasing dilutions of 4 human viruses in human serum, even human herpesvirus 5, but missed all herpesviruses in a serum-based multiplex reagent containing 11 human viruses. Unfortunately, we do not know the exact copy numbers of viruses in the samples but know that the Cq values of the herpesviruses in the multiplex reagent were ~ 30 and similar to the highest dilution of the spiked-in samples. However, all laboratories performed worse on the multiplex sample than on the other samples, which may be the result of the large number of different viruses in this sample, and the fact that the multiplex sample did not closely approximate a real clinical sample. 25 As this VMR was marketed primarily by NIBSC as a PCR control reagent, the quality of the material may not be as high as in the multiplexed sample sold specifically for metagenomic control purposes, in which we detected all herpesviruses. We also had several false-positive hits in the ring trial. One of them, the parvovirus NIH-CQV, was shown to be a common contaminant of silica spin columns used for nucleic acid extractions. 46 Others were most likely carryover contaminations by high viral loads in other samples, such as the poliovirus, which was present in high numbers in the undiluted spiked-in samples. It is difficult, if not impossible, to avoid this contamination in NGS. Although negative controls, such as the porcine lung tissue that we co-sequence in every run, may help, contamination may not be evenly distributed over the samples and not necessarily picked up by the control. Therefore, multiple factors such as number of reads, genome coverage, reads distribution over the entire genome, and anamnestic data must be considered, and data carefully interpreted to avoid false-positive reports. An important conclusion from our interlaboratory proficiency test was that it is crucial for data interpretation and reporting to know at least to some degree the origin and clinical background of the sample to be sequenced—and in the case of veterinary samples, the host species and material. Future ring trials should therefore ideally be based also on “true” clinical samples. In addition, serum samples, as used here, are not an ideal sample material for virus detection and—at least in veterinary virology—are rarely used.
Given the relatively high costs for NGS, screening pooled samples for the spectrum of viruses present may be a cost-efficient way for virus surveillance or virus profiling of a group of animals. Representative sample collection is widely used for Salmonella screening of poultry flocks, and differences in the suitability of pooled sample material have been observed; in the case of cage production units, dust was most sensitive for detection of Salmonella, whereas for free-range flocks, analysis of feces was more sensitive.4,10 Porcine viruses (e.g., circoviruses and adenoviruses) were even detected in water samples separated from manure 48 ; screening of collective sewage samples is broadly used to monitor the occurrence of enteric viruses such as norovirus or HEV in the human population.22,32 Our small pilot study showed that the same enteric viruses were found in a naturally pooled fecal sample from the pen floor and in samples from individual animals. Chewing rope liquid has been shown to be a suitable material for the specific detection of non-enteric viruses such as swine IAV or porcine reproductive and respiratory syndrome virus (Betaarterivirus suid).43,44 In the chewing rope fluid, we identified most of the enteric viruses that were also present in feces. In addition, we found 2 viruses (i.e., porcine cytomegalovirus and porcine circovirus) with a relatively high number of reads and coverage that were otherwise only identified in serum from individual animals and with just a few reads. Both viruses, together with rotavirus C, which had been identified only in the pen floor fecal sample, might have been missed if only the samples from the individual animal were sequenced. Particularly in the case of fecal samples, accumulation of intermittently shed viruses on the floor of the bay may enhance detection of enteric viruses. However, we did not sequence the samples from the single animals individually, and we did not check our results with specific tests (e.g., rtPCR). Furthermore, only a single pig pen was analyzed. However, in addition to gaining important experience in using pen floor samples for NGS, we concluded that the “naturally pooled” samples resulted in nearly the same spectrum of viruses as obtained from pooled samples of individual animals. Exceptions were the unassigned CRESS viruses and TTSuV, which were mainly detected in serum and BAL. At least for TTSuV, it is known that this virus causes persistent and long-lasting viremia without being efficiently shed, 29 which explains its absence in the pen floor samples. Even though more comprehensive studies are required, our results indicate that the combination of chewing rope fluid and pen floor fecal samples may represent a noninvasive and relatively inexpensive way to screen pig pens for circulating viruses.
The successful analysis of a broad range of clinical sample material known to be positive for a wide range of viruses was an important step in the establishment of ViroScreen. First experiences in a diagnostic setting showed successful application for virus identification, characterization, and screening of groups of animals. However, in addition to the remaining issues, such as our inconsistent results with detecting herpesviruses, the fast evolution of NGS platforms and respective reagents require constant revision of the protocols. Even more important, databases used for reference-based alignments need to be updated regularly given that the number of newly detected viruses is growing fast.
Another challenge is the interpretation of data. In contrast to specific testing, metagenomic analysis may reveal a whole list of viruses present in a sample, often with unknown causative correlation or clinical meaning. Therefore, data should be interpreted carefully, including all available factors such as number of reads, the genome coverage, distribution of reads over the entire genome, and anamnestic information, and may require additional, alternative methods to confirm results and provide causative correlation.
Compared to other methods used by diagnostic laboratories, metagenomic NGS is a relatively slow method. In our setting, the minimum time necessary from sample arrival to the final report is ~ 3 d. Nevertheless, comparing the advantages and challenges of using NGS in veterinary medicine, our ViroScreen protocol appears to be a promising tool for diagnostic laboratory and research purposes, providing comprehensive information and insights into viral diseases in animals.
Supplemental Material
sj-pdf-1-vdi-10.1177_1040638720982630 – Supplemental material for Implementation of next-generation sequencing for virus identification in veterinary diagnostic laboratories
Supplemental material, sj-pdf-1-vdi-10.1177_1040638720982630 for Implementation of next-generation sequencing for virus identification in veterinary diagnostic laboratories by Jakub Kubacki, Cornel Fraefel and Claudia Bachofen in Journal of Veterinary Diagnostic Investigation
Footnotes
Acknowledgements
We thank Prof. Mathias Ackermann for his helpful input and Prof. Xaver Sidler and Dr. Robert Graage from the Division of Swine Medicine for providing the pig pen samples.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Our work was supported by the University of Zurich and the Swiss Federal Food Safety and Veterinary Office.
Supplementary material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.