
Abstract
Quantitative real-time polymerase chain reaction (qPCR) assays were developed and validated in combination with enrichment culture for the detection and discrimination of Erysipelothrix rhusiopathiae and other Erysipelothrix species from tissue samples. The targets for SYBR green qPCR assays were the 16S ribosomal RNA gene for Erysipelothrix species and a gene involved in capsular formation for E. rhusiopathiae. The specificity of the assays was assessed with Erysipelothrix species and other related bacterial species. The limit of detection was found to be 5 colony-forming units per reaction. Amplification of DNA extracted from spleen and joint samples spiked with increasing quantities of Erysipelothrix cells was shown to be equally sensitive to DNA extracted from a pure bacterial culture. The assays were evaluated with 88 tissue samples from 3 experimentally infected pigs and 50 mice and with 36 tissue samples from 3 naturally infected pigs and 11 noninfected pigs. Results were compared with those of direct qPCR and conventional culture. The qPCR after enrichment increased the diagnostic sensitivity over that of culture and qPCR, thereby significantly reducing the total time taken for the detection of E. rhusiopathiae and other Erysipelothrix species. Therefore, this technique could be used for practical applications.
Erysipelothrix rhusiopathiae is a small Gram-positive rod bacterium that causes erysipelas in swine and a variety of diseases in other animals, as well as erysipeloid, a skin disease of humans. Swine erysipelas, a disease causing enormous economic losses in pig production, can occur as an acute septicemia or chronic polyarthritis, lymphadenitis, and endocarditis. 14 The genus Erysipelothrix was once thought to be represented by only 1 species E. rhusiopathiae, which was classified into 25 serovars on the basis of peptidoglycan antigens of the cell wall. 11 To date, the genus contains at least the following 2 species: E. rhusiopathiae, including serovars 1a, 1b, 2a, 2b, 4–6, 8, 9, 11, 12, 15–17, 19, 21, and type N, and Erysipelothrix tonsillarum, including serovars 3, 7, 10, 14, 20, 22, and 23. Serovars 13 and 18 are unclassified but are considered to be genetically distinct from the above 2 species. 11 Erysipelothrix rhusiopathiae are pathogenic for swine and chicken, but the other Erysipelothrix species are nonpathogenic. Additionally, E. tonsillarum and 2 unclassified species are isolated not only from healthy pigs but also from diseased pigs (arthritis, lymphadenitis). 10,12
Human infections with E. rhusiopathiae are typically obtained during contact or handling of animal tissues; an existing wound or injury sustained during animal handling is the point of entry. Therefore, swine erysipelas continues to be a reason for carcass condemnations at slaughterhouses, which causes economic loss to swine producers worldwide. 1,6,15 Diagnosis of the disease is mainly carried out by cultivation and identification of E. rhusiopathiae on the basis of growth and biochemical characteristics; however, the method is laborious and time-consuming. 2,10 Furthermore, it is difficult to distinguish E. rhusiopathiae from E. tonsillarum with the use of conventional diagnostic bacterial tests. 15 Therefore, a simple, accurate, and rapid method for detection of E. rhusiopathiae is highly desired. Conventional polymerase chain reaction (PCR) assays have successfully been applied to the detection of Erysipelothrix spp. A previous study 4 established a PCR assay that used primers derived from the DNA sequence encoding 16S ribosomal RNA (rRNA) of Erysipelothrix species, and another study 10 developed a PCR assay based on the DNA sequence encoding a gene involved in capsular formation in E. rhusiopathiae.
Primers for LightCycler simplex polymerase chain reaction.

GenBank accession no. AB055905.
GenBank accession no. D64177.
Recently, real-time PCR, an advanced form of PCR, has been widely used to detect bacterial agents. Compared with the traditional gel-based PCR, real-time PCR is faster and more suited to high throughput of samples and can quantitate the nucleic acid concentration. The aim of this study was to develop quantitative real-time PCR (qPCR) assays for the specific detection and discrimination of E. rhusiopathiae from other Erysipelothrix species. The experiments described herein are based on the amplification and detection of fragments of Erysipelothrix DNA coding for 16S rRNA and of fragments of E. rhusiopathiae DNA coding for a gene involved in capsular formation. The specificity and sensitivity for detection and discrimination of E. rhusiopathiae and other Erysipelothrix species in tissue samples of experimentally infected animals and naturally infected and noninfected pigs after enrichment were investigated, and the results were compared with those of conventional culture and direct qPCR.
The Erysipelothrix reference strains used in the current study included 17 strains of E. rhusiopathiae (Fujisawa, serovar 1a; 422/1E1, serovar 1b; ATCC 19414T, serovar 2; Koganei 60–0.15, serovar 2; Doggerscharbe, serovar 4; Pe'cs 67, serovar 5; Dolphin E-1, serovar 6; Goda, serovar 8; Kaparek, serovar 9; IV12/8, serovar 11; Pe'cs 9, serovar 12; Pe'cs 3597, serovar 15; Tanzania, serovar 16; 545, serovar 17; 2017, serovar 19; Bãno 36, serovar 21; MEW 22, type N), 5 strains of E. tonsillarum (ATCC 43339T, serovar 7; ATCC 43338, serovar 7; Lengyel-P, serovar 10; 2553, serovar 20; Bãno 107, serovar 22), and 2 unclassified strains in genus Erysipelothrix (Pe'cs 56, serovar 13; 715, serovar 18). Twenty-one field isolates of E. rhusiopathiae isolated from swine with arthritis or dermatitis were also used to evaluate the specificity of the qPCR. Erysipelothrix rhusiopathiae strains Fujisawa (serovar 1a), ATCC19414T (serovar 2), Koganei 65–0.15 (serovar 1a, Japanese official live vaccine strain), and Dolphin E-1 (serovar 6); E. tonsillarum strain L1–4 (serovar 7); and unclassified species strain 715 (serovar 18) were used to experimentally induce the disease in pigs and mice. Properties of the strains were described elsewhere. 5,13
The 15 other bacterial species found commonly in swine were used to determine specificity of the qPCR: Actinomyces equi, Actinobacillus suis, Actinobacillus pleuropneumoniae, Bordetella bronchiseptica, Clostridium perfringens, Arcanobacterium pyogenes, Corynebacterium suis, enterohemorrhagic Escherichia coli, Haemophilus parasuis, Pasteurella multocida, Salmonella choleraesuis, Staphylococcus aureus, Streptococcus suis, Streptococcus faecalis, and Mycoplasma hyopneumoniae.
DNA was prepared with a DNA extraction kit a as previously described. 3 The sequences of Erysipelothrix genus-specific qPCR (EqPCR) primers (E16S-F and E16S-R) targeting the 16S rRNA gene specific for Erysipelothrix species were designed on the basis of the 16S rRNA sequences of E. rhusiopathiae (GenBank accession nos. AB055905, EF494749, and EF050044), E. tonsillarum (GenBank AB034201 and AB055906), and unclassified strains of serovars 13 and 18 (GenBank AB055907 and AB055908, respectively). The sequences of E. rhusiopathiae-specific qPCR (ErqPCR) primers (Er1-F and Er2-R) targeting a gene involved in capsular formation in E. rhusiopathiae were designed on the basis of the nucleotide sequence specific for E. rhusiopathiae chromosomal DNA (GenBank D64177). The primers utilized in this study are shown in Table 1. The real-time PCR methods were designed for a simplex PCR so that all Erysipelothrix species would be detected by the EqPCR assay and that only E. rhusiopathiae would be detected by the ErqPCR assay.
The reaction volume for each individual qPCR was 20 μl, containing 10 μl of a Hot-start SYBR II Premix Ex Taq, b 0.5 μM of each primer, and 5 μl DNA template. Amplification was performed with a thermal cycler c according to the following program: an initial denaturing step at 95°C for 30 sec and 40 cycles of denaturation at 95°C for 15 sec, annealing at 58°C for 17 sec, and extension at 72°C for 20 sec. Fluorescence increase (i.e., creation of specific product) was measured during the annealing step at 58°C.
A melting curve analysis was performed after the last amplification cycle with 95°C for 0 sec, 65°C for 15 sec, and 95°C for 0 sec. Temperature transition rates were 20°C/sec for all but the last step, wherein the rate was 0.1°C/sec. A sample was regarded as positive for Erysipelothrix species when the crossing point (Cp) value of EqPCR was 36 or less and the qPCR product produced a characteristic melting curve with a discernible peak at 86.26 + 0.15°C. A sample was regarded as positive for E. rhusiopathiae when the Cp value of EqPCR was 36 or less, the fluorescence intensity by ErqPCR was 0.5 fluorescence units or more, and 2 characteristic melting curves were produced with discernible peaks at 86.26 + 0.15°C and 76.75 + 0.07°C in the melting curve screen.
To determine the limits of detection, linear ranges, and amplification efficiencies of the qPCR assays, 2 types of standard curves were constructed with the use of DNA extracted from a pure bacterial culture and DNA extracted from spleen and joint samples spiked with Erysipelothrix cells. Bacterial concentrations were determined by titration on appropriate broth and agar as described elsewhere. 13 Aliquots from dilutions (103–108 colony-forming units [CFU]/ml) were pelleted by centrifugation, and the bacterial cells were resuspended in phosphate buffered saline or the supernatants prepared from spleen and joint samples (see below). No-template controls and nonspiked samples were included in all qPCR runs.
DNA extracted from a pure bacteria culture was diluted to a concentration equivalent to 105 CFU/ml and stored in small aliquots at −20°C. To determine inter- and intra-assay variation, an aliquot was thawed and run in quintuplicate in 5 consecutive runs of the qPCR assays.
To induce an acute septicemia model of erysipelas, 3 specific pathogen-free pigs were inoculated intradermally with 1.2 × 106 CFU of Fujisawa strain, a Japanese official challenge strain for the assay of vaccine efficacy, and 10 female ddY mice d were each inoculated subcutaneously with 1.5 × 102 CFU of Fujisawa strain or 2.3 × 102 CFU of ATCC 19414 strain. During postmortem examination of the euthanized pigs, organs (lung, liver, spleen, kidney, and lymph nodes) were isolated. In addition, 5 female ddY mice d were each inoculated subcutaneously with 2.3 × 102 CFU of L1–4 strain (E. tonsillarum, serovar 7) or 4.7 × 102 CFU of 715 strain (unclassified species, serovar 18). During postmortem examination of the euthanized mice, spleens were isolated. Small pieces of isolates were inoculated on brain heart infusion (BHI) agar e and broth e (pH 7.6) supplemented with 0.1% Tween 80, f 0.3% Tris-hydroxymethyl aminomethane, g crystal violet (5 μg/ml), g and 0.03% sodium azide g for isolation and selective enrichment. Fifty milligrams of each tissue sample was finely chopped and submitted to DNA extraction.
To induce a chronic model of erysipelas, 10 female ddY mice d were each inoculated subcutaneously with 1.0 × 107 CFU of Koganei 60–0.15 strain or 2.7 × 102 CFU of Dolphin E-1 strain, which have been used to induce arthritis in mice. 4,5 After 7 days, stifle joints and spleens were isolated. The joints were finely chopped and vortexed with 3 ml of 0.85% NaCl solution. The spleens were finely chopped and homogenized in 3 ml of 0.85% NaCl solution. The supernatants (100 and 500 μl) were cultured in the BHI agar and broth for isolation and selective enrichment. After incubation, the Erysipelothrix-like colonies were Gram-stained and examined microscopically. In parallel, the supernatants and the enriched broth cultures were subjected to DNA extraction for the qPCR. The animals used in this study were cared for in accordance with regulations of animal experimentation of Nippon Institute for Biological Science, which conform to the standard principles of laboratory care.
The specificity of the assays was further validated with spleens from 3 pigs naturally infected with E. rhusiopathiae and organs (lung, spleen, and lymph node) from 11 pigs naturally infected with other bacterial agents or virus agents. Tissue samples were cultured and DNA was extracted as described above, and colonies with characteristics resembling Erysipelothrix species were Gram stained and examined microscopically. Suspected Erysipelothrix colonies were then identified as E. rhusiopathiae by PCR with the use of E. rhusiopathiae-specific primers ER1 and ER2. 10
To determine the effect of enrichment on detection sensitivity of the qPCR, the supernatants (0.5 ml) prepared from spleens and joints with known concentrations were added to the enriched broth (4.5 ml) to produce broth cultures containing 4 × 100 and 4 × 101 CFU/5 ml, respectively. The enrichment cultures were incubated at 37°C for 24 hr, and 1-ml samples of the enrichment culture were removed at intervals of 6, 9, 12, 16, and 24 hr and processed for extraction of genomic DNA.
The individual primers were analyzed by BLAST against GenBank, DDBJ, and EMBL nucleotide databases to access the specificity of the assays in silico. A set of primers for EqPCR was designed to target a 202-bp region of the 16S rRNA gene specific for Erysipelothrix strains, and another set of primers for ErqPCR was designed to target a 119-bp region of the sequences of chromosomal loci involved in capsular formation in E. rhusiopathiae. The annealing temperatures and primer concentrations of the assays were optimized to determine the temperature and primer concentration that gave the best specificity without reduction in yield. The optimal annealing temperature and primer concentration for the qPCR assays were determined to be 58°C and 500 nM, respectively.
The specificity of the assays was validated with 17 E. rhusiopathiae strains, 5 E. tonsillarum, the 2 unclassified Erysipelothrix strains serovars 13 and 18, and 21 field isolates of E. rhusiopathiae. The primers targeting the 16S rRNA gene were found to be specific to Erysipelothrix DNA, whereas the primers targeting a gene involved in capsular formation were specific to E. rhusiopathiae DNA. The use of the latter primers did not result in nonspecific reactions with DNA from the other Erysipelothrix species. Erysipelothrix rhusiopathiae contained both targets and generated the 202- and 119-bp products, but other Erysipelothrix species contained 1 target and thus generated only the 202-bp product. The specificity of the assays was also verified empirically with 15 other bacterial species. The absence of nonspecific products and primer dimmers was confirmed by melting curve analysis and gel electrophoresis
Melting curves and melting peaks of the 2 quantitative polymerase chain reaction (qPCR) assays. Melting curve analysis of the PCR products from individual PCR reactions revealed that Erysipelothrix rhusiopathiae had single peaks at 86.26 + 0.15°C and 76.75 + 0.07°C (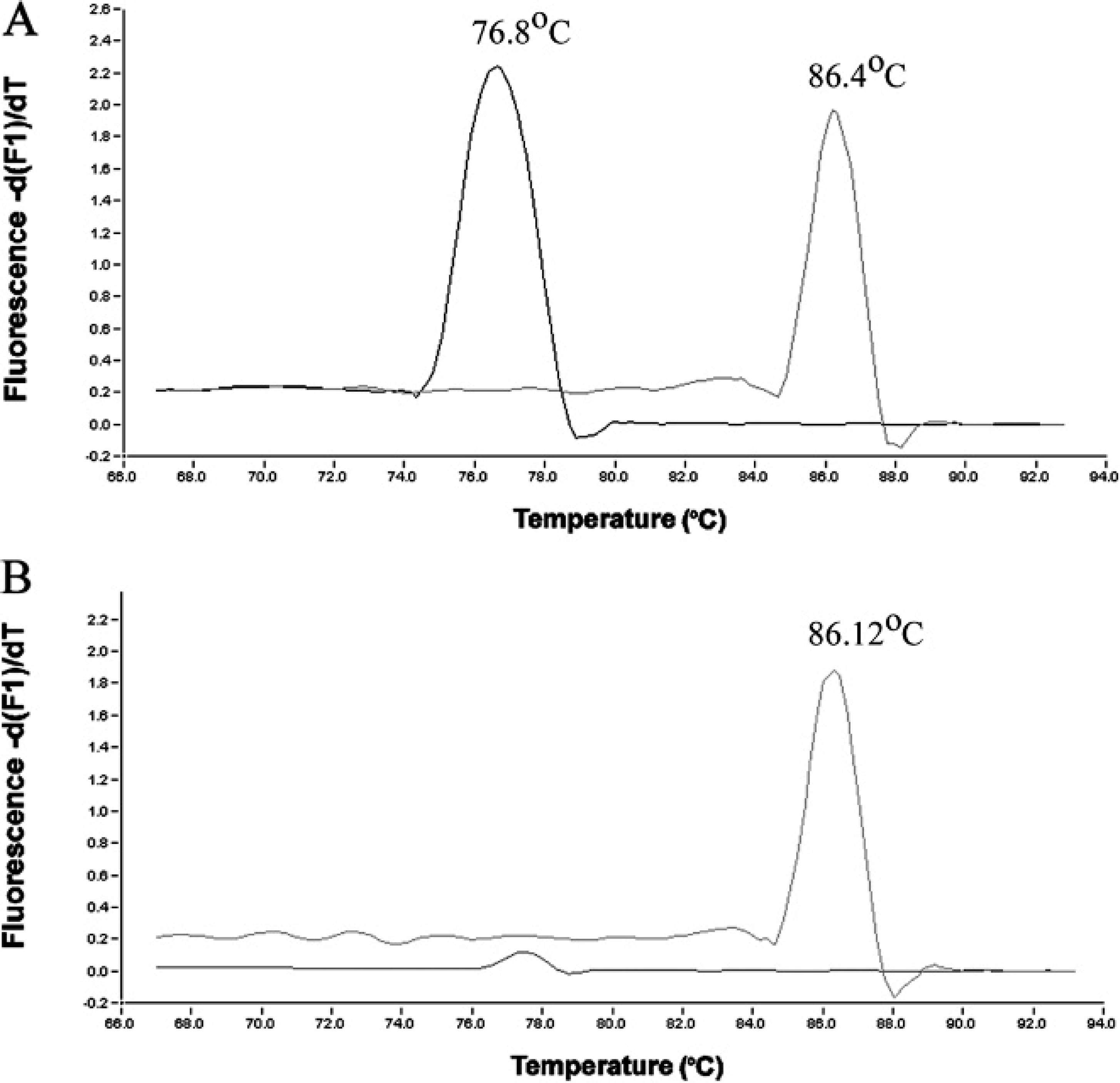
Linear dynamic ranges of 103–108 CFU/ml and 105–108 CFU/ml were observed constantly for the EqPCR and ErqPCR, respectively, with the use of DNA extracted from pure bacterial culture and DNA extracted from spleen and joint samples spiked with Erysipelothrix cells. The efficiency of the EqPCR was 0.95 + 0.05 (R 2 > 0.99) with a linear dynamic range over 6 log units (103–108). The efficiency of the ErqPCR was 0.79 + 0.03 (R 2 > 0.99) with a linear dynamic range over 4 log units (105–108). The sensitivity of the EqPCR and ErqPCR was determined to be approximately 5 CFU/reaction.
The control templates containing 105 CFU/ml were assayed in quintuplicate in both assays to determine the inter- and intra-assay variability. As determined from the Cp values obtained from 5 consecutive runs, the interrun variation for this control sample was 0.39 (average Cp, 23.8; coefficient of variation [CV], 1.65%) and 0.64 (average Cp, 30.94; CV, 2.1%) for the EqPCR and ErqPCR, respectively. The mean of the intra-assay variability was 0.32 (range: 0.28–0.42) and 0.47 (range: 0.33–0.64) for the EqPCR and ErqPCR, respectively. Results of qPCR after enrichment for the detection of Erysipelothrix strains in the tissue samples were compared with those of conventional culture and direct qPCR (Table 2).
With cases of the acute septicemia model of erysipelas, 18 pig tissue samples (including 3 each of lung, liver, spleen, tracheobronchial lymph node, mesenteric lymph node, and inguinal lymph node) and 20 mouse spleen samples were positive by qPCR, culture, and qPCR after enrichment. In addition, 10 spleen samples from mice infected with serovar 7 (E. tonsillarum) and serovar 18 (unclassified species) were positive by EqPCR but negative by ErqPCR. The Cp values in the EqPCR assay for the tissue samples (∼50 mg) were between 19.9 and 23.1, with quantitative results being 3.7 × 105–2.4 × 107 Erysipelothrix cells. These results demonstrated that tissue samples collected from animals of the acute septicemia model contained large numbers of Erysipelothrix cells.
Summary of the results of conventional culture, direct quantitative polymerase chain reaction (qPCR), and qPCR after enrichment for the detection of Erysipelothrix rhusiopathiae (ErqPCR) and other Erysipelothrix species (EqPCR) from animal tissues.
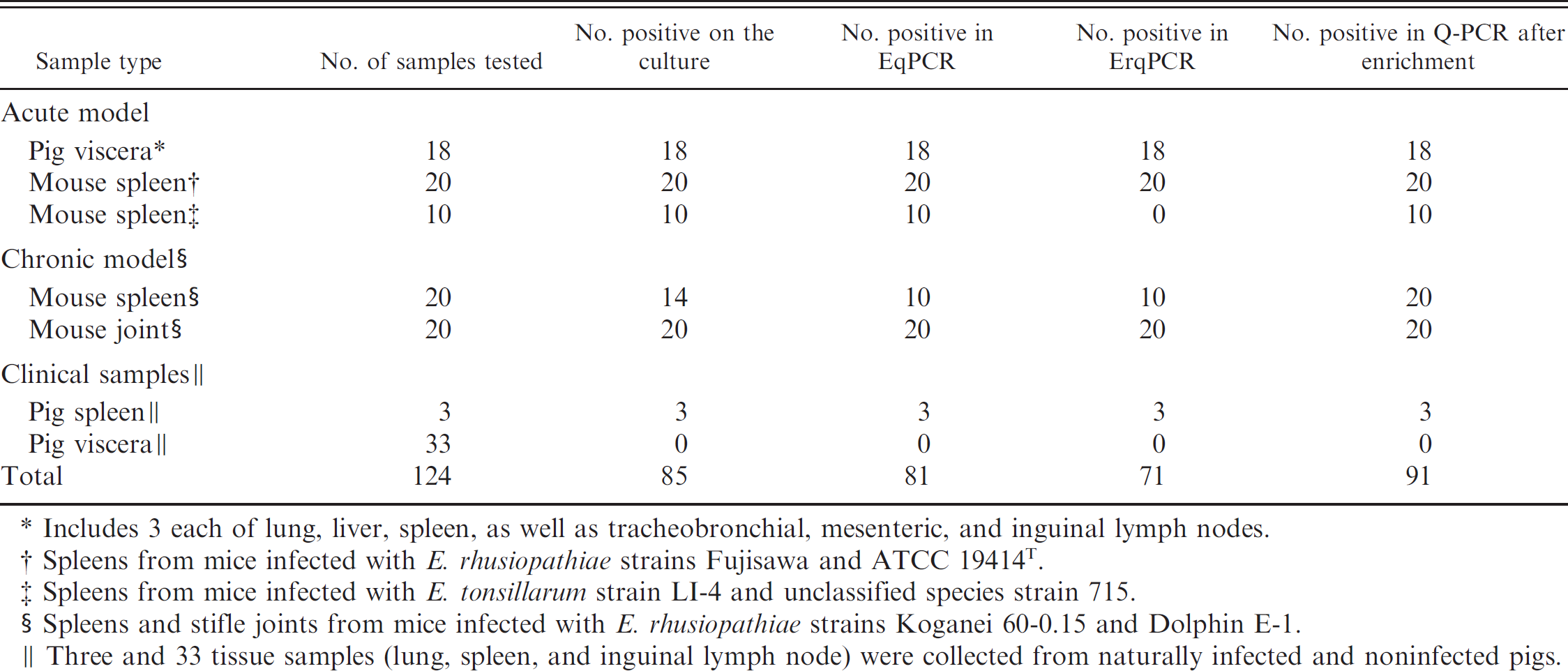
Includes 3 each of lung, liver, spleen, as well as tracheobronchial, mesenteric, and inguinal lymph nodes.
Spleens from mice infected with E. rhusiopathiae strains Fujisawa and ATCC 19414T.
Spleens from mice infected with E. tonsillarum strain LI-4 and unclassified species strain 715.
Spleens and stifle joints from mice infected with E. rhusiopathiae strains Koganei 60–0.15 and Dolphin E-1.
Three and 33 tissue samples (lung, spleen, and inguinal lymph node) were collected from naturally infected and noninfected pigs.
With cases of the chronic arthritis model of erysipelas, 20 joint samples were positive by qPCR, culture, and qPCR after enrichment. Of 20 mouse spleen samples, 10 were positive by qPCR, 14 were positive by culture, and 20 were positive by enrichment PCR assay. The joint samples had total viable bacterial counts ranging from 2.8 × 103 to 4.4 × 104 CFU/ml. The spleen samples had total viable bacterial counts ranging from 1.0 × 104 to 1.2 × 104 CFU/ml. Four samples were culture positive/qPCR negative and had total viable bacteria counts ranging from 1.0 × 101 to 2.7 × 102 CFU/ml. The Cp values in the EqPCR for the PCR-positive tissue samples were between 26.7 and 34.6. The data demonstrated that the number of E. rhusiopathiae bacteria in spleen samples was lower than those in joint samples. Comparison of the viable counts and theoretical numbers of cells showed that the number of cells obtained by qPCR were higher than the colony-forming units obtained by the viable counts in some of samples positive by both culture and qPCR (data not shown).
Three spleen samples from pigs naturally infected with E. rhusiopathiae tested positive by qPCR, culture, and qPCR after enrichment. The Cp values in the EqPCR assay for these samples (∼50 mg) were between 31.8 and 33.1, with quantitative results being 1.6 × 103–3.4 × 103 Erysipelothrix cells. However, 33 tissue samples (11 each of lung, spleen, and inguinal lymph node) from pigs naturally infected with infectious agents other than E. rhusiopathiae were negative by all 3 methods.
The qPCR assays consistently produced strong amplification after a 9- and 12-hr incubation for the original broth cultures containing 4 × 101 CFU/5 ml and 4 × 100 CFU/5 ml, respectively. The data showed that by incorporating an enrichment period of 12 hr, the qPCR assays allowed detection of 8 CFU/ml of the original sample.
The genus Erysipelothrix contains 2 main species, E. rhusiopathiae and E. tonsillarum, and 2 unclassified species. Erysipelothrix rhusiopathiae are pathogenic for swine and chicken, but other Erysipelothrix species are not. Additionally, E. tonsillarum and the 2 unclassified species are isolated not only from healthy pigs 10 but also from pigs with arthritis or lymphadenitis caused by other bacterial infections. 12,14 Therefore, the accurate and rapid discrimination of these closely related species is very useful not only for meat inspection centers but also for clinical diagnosis.
In the current study, the real-time qPCR assays for the detection and discrimination of E. rhusiopathiae and other Erysipelothrix species from tissue samples after enrichment were developed, with the results being compared with those of conventional culture and direct qPCR. The main advantages of the real-time PCR assay over the traditional PCR test were that 1) it is performed in a closed system in which post-PCR handling is not required, which reduces assay labor, material costs, and amplicon contamination; 2) the assay is highly specific and sensitive, and product information can be monitored in real time, making quantification of bacteria possible; and 3) it shortens the turnaround time for results and has the potential for automation, which could be very useful for examining a large number of samples in a short period of time.
The EqPCR assay from DNA extracted from the pure bacterial culture in this study demonstrated a linear range of quantification over 6 orders of magnitude and a quantitative limit of detection of approximately 5 CFU, whereas the ErqPCR assay had a linear range of quantification over 4 orders of magnitude and a detection limit of approximately 5 CFU/reaction.
The qPCR assays after enrichment reported herein were applied to the quantitative detection and discrimination of E. rhusiopathiae and E. tonsillarum in DNA extracts from 88 tissue samples of experimentally infected animals and from 3 and 33 tissue samples from naturally infected and noninfected pigs, respectively. Eighty-five of the 124 samples were found positive by culture; 81 and 71 samples were found positive by direct EqPCR and ErqPCR, respectively; and 91 and 81 samples were found positive by EqPCR and ErqPCR after enrichment, respectively. Five spleen samples each from mice infected with serovars 7 (E. tonsillarum) and 18 (unclassified species), whose genomic DNA harbor only a target (16S rRNA), were positive by EqPCR but negative by ErqPCR. Four spleen samples from mice developing arthritis positive by culture and negative by direct qPCR were thought to have a number of template molecules below the detection limit by qPCR. Low levels of bacteria are common in samples from animals with chronic erysipelas. 10,15 As expected, 3 spleen samples from pigs infected with E. rhusiopathiae were positive by qPCR, culture and qPCR after enrichment, whereas a total of 33 samples from naturally noninfected pigs were negative by all 3 methods. All samples that were culture positive were also qPCR positive after enrichment; however, qPCR after enrichment detected an extra 6 positives (Table 2). It is possible that the number of bacteria in these samples was on average low and close to the detection limit of direct culture. Correlation was good between direct culture and enrichment qPCR. The qPCR detection of E. rhusiopathiae was better after 12 hr of selective enrichment culture.
Quantitative PCR results were compared with total viable Erysipelothrix counts for the samples tested. When Erysipelothrix cultures were used, correlation between the qPCR method and the conventional culture method agreed with previous findings. 7,16 When the original samples were tested, qPCR results yielded higher values in some of the samples, in agreement with previous reports. 8,9 The results of the current study appear plausible because amplification of Erysipelothrix cultures occurs with the majority of cells in a viable cultivable state. Thus, qPCR and culture values should coincide. When tissue samples were tested, all DNA was amplified by qPCR, including DNA from dead bacteria. Colony counts were performed on selective agar, but if some of the cells were dead or injured, this would prevent their recovery on selective agar. For these reasons, the number of cells calculated by qPCR is higher than the number of colony-forming units.
Some studies have described qPCR assays that detect directly pathogenic organisms from clinical samples. However, the application of such methods for detection of E. rhusiopathiae appears to be difficult because low levels of the bacteria are common in samples from animals with chronic disease. Therefore, the recovery of Erysipelothrix species from human, veterinary, and environmental samples usually requires a 2-stage process of enrichment in broth followed by plating onto agar media. The method is advantageous because cultivation of samples from animals in enrichment broth is a routine practice in diagnostic laboratories, and the bacteria obtained by enrichment are sometimes used for further analyses. Traditional PCR assays are also used for the detection of Erysipelothrix cells after enrichment. 2,10,15 These PCR assays require an enrichment period of 24 hr and a detection period of at least 6 hr and have a detection limit of approximately 103–105 CFU/reaction, whereas qPCR requires an enrichment period of 12 hr and a detection period of 90 min (including 35 min for DNA extraction and approximately 55 min for the qPCR), with the results being available immediately at the end of PCR cycling. The real-time PCR described here offers a fast tool with high sensitivity and specificity for the detection of and discrimination between E. rhusiopathiae and other Erysipelothrix species from tissue samples, and therefore could be useful for implementation in meat inspection centers and diagnostic laboratories.
Acknowledgements. The authors thank Fumiya Kawahara for his advice in real-time PCR experiments.
Footnotes
a.
High Pure PCR Template Preparation Kit, Roche Diagnostics GmbH, Mannheim, Germany.
b.
Takara Bio Inc., Otsu, Shiga, Japan.
c.
LightCycler®350S, Roche Diagnostics GmbH, Mannheim, Germany.
d.
Female ddY mice, Nippon SLC, Shizuoka, Japan.
e.
Difco Laboratories Inc., Sparks, MD.
f.
Kanto Chemical Co. Inc., Tokyo, Japan.
g.
Nacalai Tesques Co. Inc., Kyoto, Japan.