
Abstract
The successful eradication of pseudorabies in U.S. domestic swine was accomplished through the use of glycoprotein E (gE) deleted modified live virus vaccines and an accompanying gE differential enzyme-linked immunosorbent assay (ELISA). Yet, pseudorabies virus (PRV) was established in feral swine in the United States, becoming a potential reservoir of PRV for infection of domestic swine and other native wildlife. A critical need for the current PRV surveillance program in the United States is the rapid detection of PRV infection. For this reason, a set of 2 real-time polymerase chain reaction (PCR) assays by using TaqMan chemistry was developed and evaluated for their capability in the detection and differentiation of field and vaccine strains of PRV. PCR primers and probes were designed for gB and gE genes of PRV, respectively. The newly developed PRV-specific real-time PCR assays could detect all wild-type PRV isolates from diagnostic submissions and differentiate them from vaccine strains. The analytical sensitivity of the assays was approximately 0.1 plaque-forming units per reaction. The assays were highly specific for PRV, because no positive results were obtained from testing other common swine viral pathogens and other animal herpesviruses. The results of testing samples from domestic and feral swine and from bovine showed that the real-time PCR assays are more sensitive than gel-based PCR. These results demonstrated the potential application of the developed real-time PCR assays as a differential test for rapid and specific detection of PRV in domestic and feral swine, as well as nonporcine species that can be infected with PRV and serve as carriers.
Keywords
Introduction
Pseudorabies, also known as Aujeszky's disease, is a disease caused by the Pseudorabies virus (PRV) (officially Suid herpesvirus 1; family Herpesviridae, subfamily Alphaherpesvirinae, genus Alphaherpesvirus). 15 Although swine are the natural host and reservoir for PRV, the virus can establish fatal infection in most mammals, with the exception of humans. 15,26 In young piglets, PRV infection is often fatal, and animals die from central nervous system disorders. In contrast, older pigs generally develop respiratory disease, survive the acute infection, and carry the virus in a latent form for their entire life. In pregnant sows, PRV infection normally leads to reproductive failure. 36
Although this disease has been eradicated from some countries, 3,15,16,22 including the United States, 26 pseudorabies epidemics still occur throughout the world, 23,28 and it is still considered a major swine disease, which causes great economic loss for affected countries. Most eradication programs were completed through the use of live virus marker vaccines that lack the nonessential glycoprotein E (gE) and an accompanying differentiating infected from vaccinated animals (DIVA) serologic test (i.e., gE differential enzyme-linked immunosorbent assay [ELISA]). 15,22,25 Even though pseudorabies has been eradicated from U.S. domestic swine by using this technology, 26 there are still 2 major obstacles to maintaining a PRV-free status: feral swine and improved diagnostics.
In the United States, PRV is well established in feral swine, 6,27,30 which serve as a potential reservoir for infection of domestic swine and other native wildlife species. Normally, feral swine are subclinically infected with PRV, and the strains isolated from feral swine are markedly attenuated in domestic swine. 10 However, domestic swine can be infected with PRV via contact with feral swine as found recently in Wisconsin (Wisconsin responds to pseudorabies cases: June 15, 2007, JAVMA News. Available at: http://www.avma.org/onlnews/javma/jun07/070615r.asp. Accessed Feb. 22, 2008). This case reinforces the need to have rapid and sensitive PRV diagnostic methods available once PRV has been introduced into naïve domestic swine.
Laboratory diagnostic tests that have been commonly used for the detection of PRV or virus infection are virus isolation (VI), fluorescent antibody tissue section (FATS) test, and serology, such as serum virus neutralization, latex agglutination test, and ELISA. 15,34 Although these methods are specific, it may take 1 to 2 weeks to obtain test results or diagnostic confirmation when VI or serologic tests are performed. In comparison with the above methods, the gel-based polymerase chain reaction (PCR) test 1,11,12,33 is a fast, sensitive, and accurate assay in which results may be available within hours. Moreover, this technique is able to detect PRV in the early stage of infection before formation of antibodies, as well as in latent infection. The real-time PCR technique has all the advantages of gel-based PCR. Furthermore, real-time PCR is more rapid and sensitive than the gel-based PCR technique. Other benefits include a minimal risk of cross-contamination because of the absence of post-PCR manipulation of the amplicon. 8,17 Real-time PCR technology has recently been described to detect PRV from swine semen 32 and from the nervous tissue of pigs. 35 However, this test was not able to differentiate wild-type PRV from the gene-deleted marker vaccine viruses.
Feral swine are becoming a potential reservoir of PRV for infection of domestic swine and other native wildlife species in the United States. A critical need for the current PRV surveillance program in the United States is the rapid detection of PRV. For this reason, real-time PCR assays by using TaqMan chemistry for gB and gE genes of PRV were developed and evaluated for general PRV detection and differentiation between wild-type PRV and vaccine virus in the current study.
Materials and methods
Viruses and cells
Eight cell-culture amplified, field PRV isolates and 5 gE-deleted commercial vaccines, along with the Shope strain of PRV (laboratory reference strain), Porcine reproductive and respiratory syndrome virus (PRRSV) VR-2332 and Lelystad strains (prototype North American and European genotypes, respectively), Porcine circovirus-2 (PCV-2), Swine influenza virus (SIV) H1N1 and H3N2, Porcine respiratory coronavirus, Transmissible gastroenteritis virus (TGEV), Porcine cytomegalovirus (PCMV; also known as Suid herpesvirus 2), Porcine teschovirus (PTV), Porcine lymphotropic herpesvirus-1 (PLHV-1), Bovine herpesvirus 1 (BoHV-1; commonly known as Infectious bovine rhinotracheitis virus), Equid herpesvirus 1 (EHV-1), Equid herpesvirus 3 (EHV-3), Rabies virus (RABV), Bovine reovirus, and Bovine adenovirus (BAdV). The porcine kidney cell line PK-15 a was used to propagate PRV and was maintained in Dulbecco Modified Eagle Medium (DMEM) b supplemented with 10% fetal bovine serum, b penicillin (100 IU/ml), and streptomycin (100 μg/ml). b The vaccine viruses were used directly out of the bottle, without any further propagation in cell culture.
Tissues and nasal swab samples
Thirty-eight tissue archival samples (brain, tonsil, heart, spleen, and lung) from 16 confirmed PRV field cases, 1 gE-deleted PRV vaccine (Marker Gold, Schering-Plough), and 1 reference PRV (Shope strain) were used in the present study. The tissue samples were collected from 1992 to 2002 and were confirmed positive for PRV by VI at that time. One sample was a bovine brain sample, and the rest were of swine origin.
In addition, 36 tonsil samples from domestic swine, which were collected among submissions to the Iowa State University Veterinary Diagnostic Laboratory (ISUVDL) as part of the Animal and Plant Health Inspection Service post-PRV eradication surveillance program, were used for the study. These samples were all tested by a FATS assay 15 at ISUVDL and were confirmed to be negative for PRV. Furthermore, 204 tonsil samples and 136 nasal swabs from feral swine were used in the study. These samples were collected as part of the Classical swine fever virus (CSFV) surveillance program of the National Animal Health Laboratory Network and confirmed to be negative for CSFV by PCR testing.
Primers and probes
Two sets of primers and probes specific to certain regions of gB and gE genes of PRV were designed by using ABI Prism Primer Express software version 2.0 c for general detection (gB) and differentiation (gE) of field and vaccine strains of PRV based on sequence information available in GenBank and the authors' sequencing efforts of the 5 commercial PRV marker vaccines. Probes were labeled with 6-carboxyfluorescein and with 6-carboxytetramethyl-rhodamine at the 5′ and 3′ ends, respectively, and purchased from a commercial vendor. c Nucleotide information of each primer or probe is summarized in Table 1. The primers for gel-based PCR were made based on a previous report. 1 In addition, 2 sets of primers for developing a plasmid standard as molecular reference were designed based on the targeted sequence of real-time PCR (Table 1).
Standard plasmid as molecular reference
Two standard plasmids as molecular references were constructed by using pCR 4-topoisomerase I (TOPO) b vector and were used to determine the detection limit of real-time PCR. Polymerase chain reaction amplicons from gE (position 592–919) and gB genes (position 461–794) were obtained by using primer pair gE592F/gE919R for gE and gB461F/gB794R for gB (Table 1) from 40 ng of genomic deoxyribonucleic acid (DNA) of the Shope strain as a template. The PCR products were ligated into pCR 4-TOPO vector by TOPO Taq-amplified (TA) Cloning Kit. b The cloned DNA was confirmed by sequencing. The cloned DNA was prepared with the Qiagen Plasmid Mini Kit d and its concentration determined by spectrophotometry. e Copy numbers of each standard plasmid were then was determined by calculating the molecular weight of each cloned plasmid (Qiagen: 2004, QuantiTect probe PCR handbook. Available at: http://www1.qiagen.com/literature/handbooks/INT/pcrlit.aspx#QTPCRpr. Accessed Feb. 22, 2008).
Primers and probes used in real-time polymerase chain reaction (PCR) assay, and primers used in standard reference plasmid construction and gel-based PCR.*
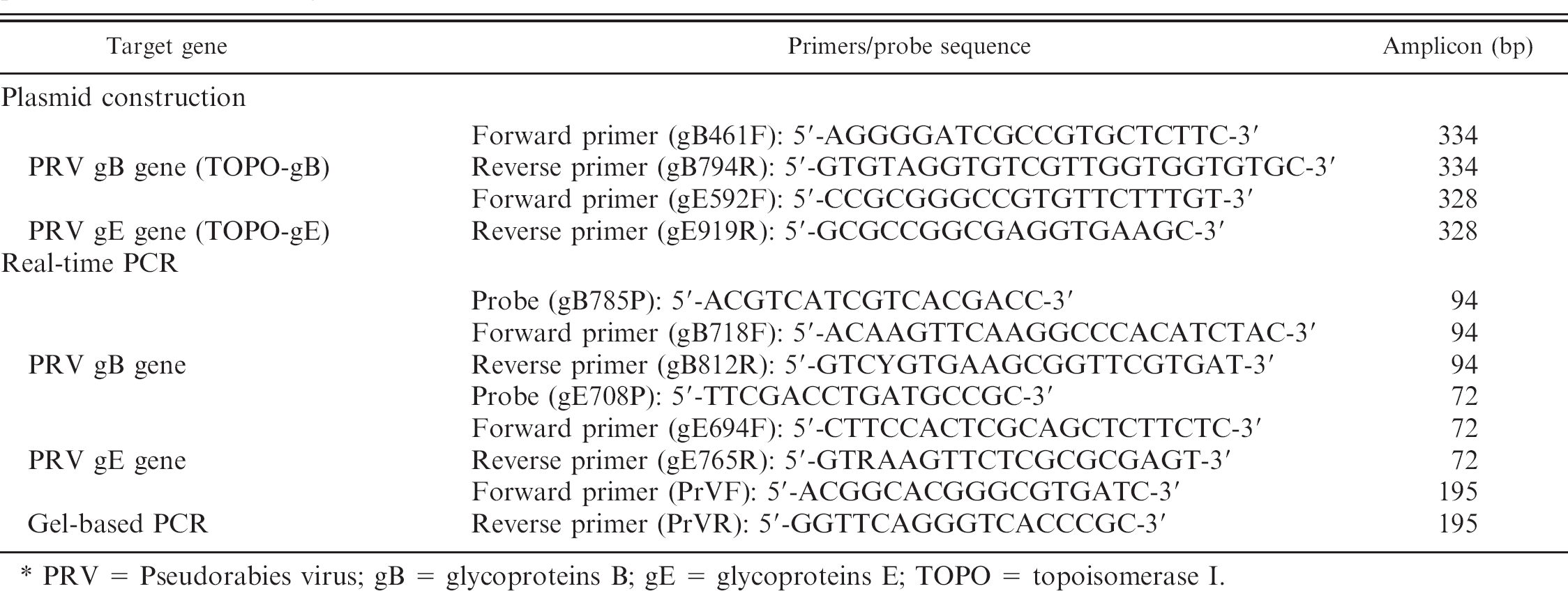
PRV = Pseudorabies virus; gB = glycoproteins B; gE = glycoproteins E; TOPO = topoisomerase I.
DNA preparation
All viral DNA was extracted from samples with the QIAamp DNA Mini Kit d by using spin column protocol as recommended by the manufacturer. In brief, 100–200 mg of minced tissue were transferred to the Eppendorf 2 ml Safe-Lock tube h that contained 5-mm stainless-steel grinding balls, and then 1 ml of DMEM media was added (i.e., 10–20% tissue homogenate). Tissue was homogenized by using Qiagen TissueLyser d for 2 min at 18 Hz according to the manufacture's instructions. The DNA was extracted from 200 μl of each cell culture fluid (i.e., isolate), vaccine, nasal-swab sample, or tissue homogenate. Viral DNA was eluted in 50 μl or 200 μl (40 blinded PRV samples from the National Veterinary Services Laboratories [NVSL], Ames, IA) of nuclease-free water, and 2.5 μl DNA extracted from cell cultures (or 10 μl from the other samples) was used as template for PCR.
PCR and sequencing
A QuantiTect probe PCR Kit d was used with a 25-μl reaction volume for real-time PCR. The assay was performed on the Smart Cycler II f by using QuantiTect probe PCR Master Mix that contained 2X QuantiTect probe PCR Master Mix 12.5 μl, template 2.5 μl (or 10 μl), gE694F or gB718F (20 μM) 0.5 μl, gE765R or gB812R (20 μM) 0.5 μl, gE708P or gB785P (25 μM) 0.2 μl, and R Nase-free water 8.8 μl (or 1.3 μl). The thermocycling condition was set as follows: 95°C for 15 min, then 40 cycles of 94°C for 15 sec and 62°C for 60 sec. Cutoffs for gB and gE real-time PCR assays were 35 Ct (threshold cycle) and 39 Ct, respectively.
The gel-based PCR assay was conducted as described previously, 1 with modifications. GoTaq Green Master Mix g was used in a volume of 25 μl that contained 2.5 μl (or 10 μl) of template, 12.5 μl of GoTaq Green Master Mix, 0.5 μl of each primer (PrVF and PrVR), and 9 μl (or 1.5 μl) of RNase-free water. The PCR condition was as follows: 94°C for 5 min, then 40 cycles of 94°C for 20 sec, 55°C for 30 sec, and 72°C 60 sec, followed by an extension of 7 min at 72°C. When necessary, PCR products were purified by using the QIAquick Gel Extraction Kit d and were sequenced by using an ABI 3730 DNA Analyzer c at the Iowa State University Nucleic Acid Facility.
Assessment of test performance
The analytic specificity of the developed gB and gE realtime PCR assays was evaluated for lack of cross reactivity with common swine viral pathogens (PRRSV, PCV-2, SIV, TGEV, PCMV, PTV, PRCV), RABV, Bovine reovirus, BAdV, and other herpesviruses (PLHV-1, BoHV-1, EHV-1, and EHV-3). The analytic sensitivity of the assays was compared with VI. Serial dilutions of the PRV Shope strain were prepared in cell-culture medium and 10 μl of each dilution inoculated onto PK-15 cell monolayers or extracted for the PCR. In addition, the detection limit of the assay was assessed by using plasmid standards prepared as described earlier. Plasmid stocks that contained 109 copies of TOPO-gB or TOPO-gE per 2.5 μl were prepared, 10-fold serially diluted with RNase-free water, and then used in the PCR. Forty PRV-positive samples from NVSL, 36 tonsil samples from domestic swine, and 204 tonsil samples and 136 nasal swabs from feral swine as mentioned above were used to assess the diagnostic performance of the developed real-time PCR compared with the results from gel-based PCR.
Virus isolation
Virus isolation for PRV in feral swine samples was performed on PK-15 cell line. 24 Briefly, 80–90% confluent monolayers of PK-15 cells were prepared in 96-well plates and then were inoculated with 100 μl of diluted PRV per well. Inoculated cells were incubated at 37°C in 5% CO2 atmosphere for 96 hr, with daily monitoring of the development of cytopathic effects. After a 96-hr incubation period, the cells were fixed by cold 80% acetone, and the presence of PRV was determined by an indirect fluorescent antibody test by using a porcine polyclonal antibody raised against PRV (NVSL) and mouse anti-pig immunoglobulin G labeled with fluorescein isothiocyanate. h Virus isolation on the archival tissue set from NVSL was not attempted a second time as part of the current study.
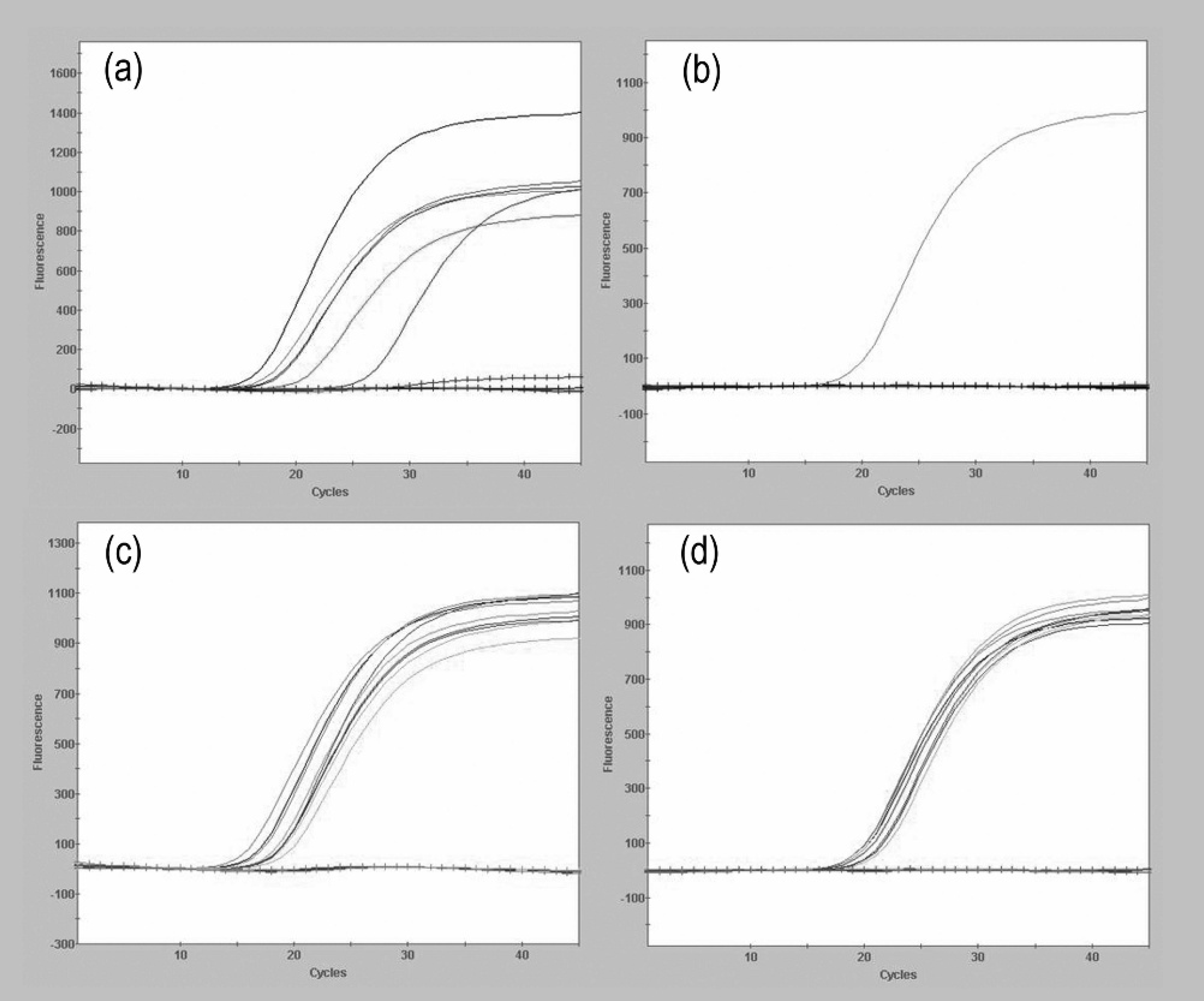
Specific detection of glycoproteins B (gB) and/or E (gE) from commercial vaccines (panels
Results
Analytic performance of gB and gE real-time PCR assays
No positive results for gB and gE were obtained from PRRSV, PCV-2, SIV (H1N1 and H3N2), PCMV, PRCV, TGEV, PTV, PLHV-1, BoHV-1, EHV-1, EHV-3, RABV, Bovine reovirus, and BAdV, which demonstrated that the PCR assays were specific for PRV. In contrast, all of the commercial marker vaccines, field PRV isolates, and the Shope strain were positive for gB (Fig. 1a, 1c). Although all the field PRV isolates and Shope strain were positive for the gE gene (Fig. 1d), all of the commercial marker vaccine viruses were negative (Fig. 1b).
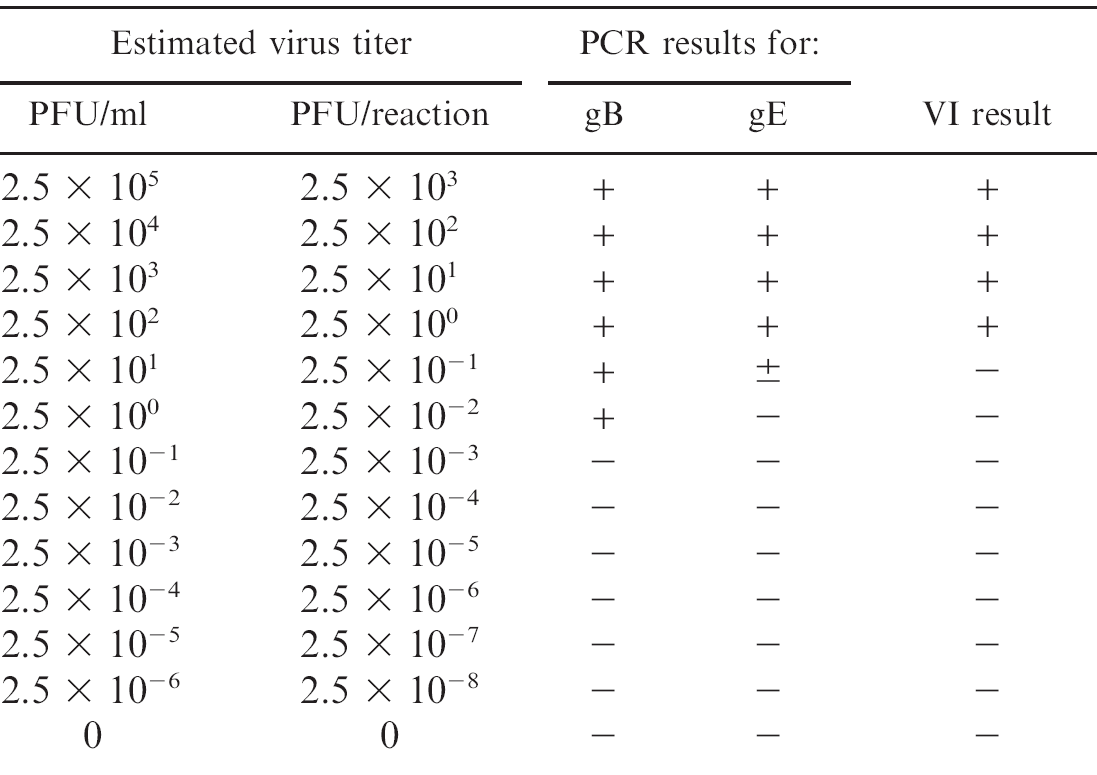
Under the reaction conditions used in the current study, it was possible to detect approximately 1 copy of the gB gene and 10 copies of the gE gene from standard plasmids of the TOPO-gB and the TOPO-gE. The log-linear standard curves generated from these studies revealed the accuracy of DNA amplification over a wide range of concentrations for both gB and gE plasmids. By using a set of serial 10-fold dilutions of the PRV Shope strain that contained a known amount of virus (plaque-forming units [PFU]), the analytic sensitivity of the PCR assays was estimated to be approximately 0.5–1 PFU per reaction for gB and gE; this sensitivity was comparable with the sensitivity of the VI (Table 2).
Diagnostic performance of gB and gE real-time PCR assays
Of the 16 PRV field cases confirmed by VI, the real-time PCR assays reconfirmed the presence of PRV (wild type) in 11 cases (68.8%). Twenty nine of the 38 tissue samples positive for PRV by VI (76.3%) were positive by both gB and gE real-time PCR assays. Twenty-six of the 29 samples (90%) that were positive by real-time PCR for gB and gE were also positive by gel-based PCR for gB gene (Table 3). These samples were retested by all of the PCR assays, and the same results were obtained. Seventeen gel-based PCR products from different field cases were randomly selected, as well as the marker vaccine and Shope strain, for sequencing; sequence analyses confirmed that all PCR products were specific for sequences of PRV. When another gel-based PCR targeting the gE gene (amplified by primer pair gE592F/gE919R) was used to evaluate these 19 gB-positive samples, 18 of the 19 samples were positive for gE, except the marker vaccine virus. All 18 gE-specific amplicons were sequenced and confirmed to be PRV sequences.
Analytic sensitivity of the real-time polymerase chain reaction (PCR) for glycoproteins B (gB) and E (gE) genes of Pseudorabies virus compared with virus isolation (VI) in PK-15 cells. The comparison was done on a set of serially diluted PRV (Shope strain) whose titer (plaque-forming unit [PFU]) was predetermined by plaque assay in PK-15 cells.
No evidence for PRV was found in any of the 36 tonsil samples collected from domestic swine by both the real-time and gel-based PCR assays. In contrast, PRV DNA was detected in 2 of 204 tonsils and 10 of 136 nasal-swab samples collected from feral swine in the southern United States (Table 4). One of the 2 tonsil samples was positive by both the gB and gE real-time PCR assays and the gB gel-based PCR. The PCR product was sequenced and confirmed to be specific for PRV. The other tonsil was positive only by the gB real-time PCR assay. One of the 10 nasal swabs was positive for PRV DNA (gB and gE) by all 3 PCR assays, 3 were positive for gB by the real-time and gel-based PCR assays, and 6 were positive only by the gB real-time PCR assay (Table 4). However, PRV was not isolated from any of the PCR-positive samples.
Summary of polymerase chain reaction (PCR) testing for glycoproteins B (gB) and E (gE) genes of Pseudorabies virus and sequencing results on 40 samples from 16 different confirmed PRV cases, which were blindly provided by the U.S. Department of Agriculture National Veterinary Services Laboratory along with known PRV (Shope strain) and commercial gene-deleted marker vaccine viruses.
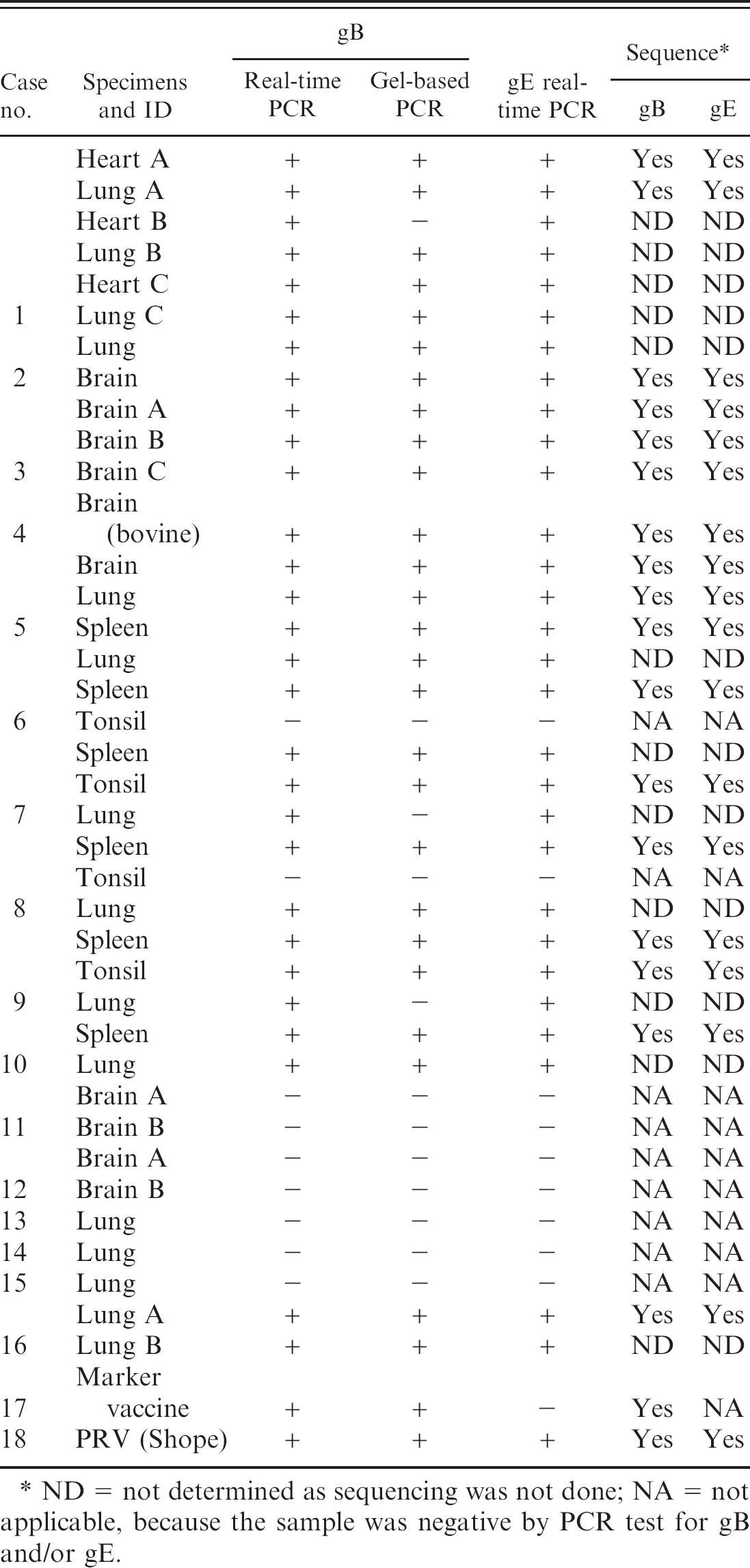
ND = not determined as sequencing was not done; NA = not applicable, because the sample was negative by PCR test for gB and/or gE.
Overall, results of the real-time and gel-based PCR assays agreed on 369 of 376 field samples (98%) as summarized in Table 4. All of the 7 discordant samples were positive by the real-time gB PCR and negative by gel-based gB PCR, which indicated that the sensitivity and specificity of the real-time PCR test relative to the gel-based PCR was 100% and 98%, respectively.
Summary of real-time polymerase chain reaction (PCR) and gel-based PCR results on samples from feral swine and domestic swine.

Number of positive animals/number of animals tested.
Discussion
The real-time PCR assays described in the current study have the ability to detect PRV and to differentiate gE-deleted vaccine viruses from wild-type PRVs. Although the use of real-time PCR for detection of PRV in boar semen 32 and in nervous tissues of pigs 35 was reported, to the authors' knowledge, this is the first description of using a real-time PCR test for rapid and specific detection and differentiation of wild-type PRV from the commercial gE-deleted marker vaccine viruses.
In the current study, the gB gene was chosen as the target gene for universal PRV detection, because it was known to be highly conserved in the PRV genome. The gE gene was used in differentiating vaccine from field PRVs, because all strains of the commercial marker vaccines were genetically engineered to delete the gE gene from the vaccine virus. Besides differential capability, the newly developed real-time PCR assays were analytically more sensitive than the gel-based PCR and VI assays. Its detection limit was estimated to be 0.1 PFU or 1 copy of the gB gene, and 1 PFU or 10 copies of the gE gene (Table 2). The reason for lower sensitivity of gE-specific real-time PCR than the gB-specific real-time PCR might be that the gE gene is not highly conserved among PRVs compared with gB gene. In addition to good sensitivity, the assays were specific for PRV, as shown when tested against other common swine viral pathogens and other herpesviruses. More importantly, both of these assays were able to detect wild-type PRV in a bovine sample, which implied that the assays could be used to test samples from nonporcine species for PRV infection. Furthermore, the same condition was used for both gB and gE real-time PCR, and the specific primers and probe of individual real-time PCR target a certain region of different genes. Therefore, it is possible to combine 2 real-time PCR assays into a multiplex real-time PCR to detect 2 signals (genes) in 1 reaction. It remains to be determined whether the sensitivity and specificity of the multiplex real-time PCR will be the same as those of the individual real-time PCR assays.
After an acute infection, PRV, like other alphaherpesviruses, is able to establish a life-long latent infection in the nervous system. The latency is a critical issue when it comes to managing the PRV-free status of the United States. There are 3 challenges to diagnosing a PRV outbreak: 1) detecting virus in a pig before seroconversion—acute phase; 2) detecting seroconversion—convalescent phase; and 3) detecting the latent phase. In general, the commercially available general (e.g., gB blocking ELISA 9 ) and DIVA (e.g., gE blocking ELISA 31 ) serologic tests are used to detect PRV-specific antibodies and to differentiate between wild-type PRV infection and vaccination during the convalescent stages. These assays are useful for PRV serodiagnosis; however, they do not detect acute PRV infections, and, in some swine, there is decay in antibody levels in the latent phase, which produces false-negative results. 13,19 In contrast to the acute and convalescent phase of infection, it is difficult to detect PRV in antemortem samples collected from latently infected pigs. Therefore, detecting latently infected swine efficiently can be a major impediment to the control and eradication of PRV. Although a series of techniques were developed to detect latent PRV infections in postmortem samples, 2,4,20 the PCR technique proved to be rapid, sensitive, and accurate. 5,7,11,18,29,33 One report described a gel-based multiplex PCR for rapid detection of PRV and other swine pathogens, 12 and, in another study, a real-time PCR was reported to be more sensitive than gel-based PCR assays. 35 This observation was confirmed in the present study.
Interestingly, samples from 5 PRV cases, which were confirmed positive by VI at the time of collection, were negative by both real-time and gel-based PCR (Table 3). The samples were lung or brain tissue collected more than 10 years ago from field cases of suspect pseudorabies. Presumably, the negative PCR test results on these tissues was likely because of a loss of viral nucleic acid over time and/or that the tissue portion selected for nucleic-acid extraction did not contain PRV DNA. The difference in sample volume between VI and PCR testing may also account for this. Primer and/or probe mismatch could be a possibility but is unlikely, because both real-time and gel-based PCRs were negative. The storage history on these tissues was not complete; hence, the potential storage effect on the stability of the viral DNA is unknown. Because of logistical concerns related to biosecurity, only one small portion of each tissue was selected for nucleic-acid extraction and was provided as extracts for the study. When testing the samples from feral swine, a few samples were identified that were PRV positive by real-time and gel-based PCR. Positive PCR results appear to be real, because the presence of PRV in these samples was confirmed to be wild-type virus by nucleic acid sequencing, and the sequence is similar to PRV known to be circulating in feral swine in the region from which the samples were collected. However, PRV was not isolated from these samples. Such a discrepancy could be attributed to the difference in sensitivity of PCR and VI assays. Storage conditions might also contribute to negative VI results, because these samples were collected as part of the national survey for CSFV in domestic and feral swine and were transported from other diagnostic laboratories to ISUVDL after being stored for a certain period of time. The observation of a loss of infectious PRV under the above conditions emphasizes that the proper storage and transportation of samples is very important to maintain the infectivity of PRV. At the same time, this observation supports the utility and value of molecular assay for diagnostic investigation and surveillance as the assay can detect both infectious and noninfectious virus material. Moreover, analysis of the feral swine samples supports the need for additional study to investigate the gB-positive/gE-negative cases detected in the current study. It is not clear if this result represents PRV vaccine derivatives 14 or a natural gE-deleted virus 21 circulating in feral swine.
The gE-deleted marker vaccine is used for eradication of PRV worldwide. Previous studies showed that PRV is well established in the U.S. feral swine population. 6,27 Data from the current study confirmed those reports, even though a few of the feral swine samples were positive for PRV DNA. Recently, PRV infection of wild pigs in Nebraska (PRV found in Nebraska feral hogs: February 12, 2007, Southwest Nebraska News. Available at: http://www.aasv.org/news/story.php?id=2283. Accessed Feb. 26, 2008), and a PRV outbreak in 2 Wisconsin domestic swine herds were reported. The latter case was likely to have had contact with feral swine as ruled by the U.S. Department of Agriculture. All these data suggest that feral swine are a potential threat for PRV infection of the U.S. national domestic herd. The newly developed real-time PCR assays reported in the current study were shown to be convenient and useful for the detection and differentiation of field PRV and vaccine viruses. Moreover, the current study demonstrated that the assays can be used to detect PRV in feral swine as well as in nonporcine species, even though further evaluation of the assays on nonporcine species is needed. Therefore, these assays may play an important role in protecting the achievements of the U.S. PRV eradication program.
Acknowledgements
The authors thank Ms. Deb Adolphson (National Animal Disease Center, ARS, USDA) for technical assistance, Dr. Karen Harmon (Veterinary Diagnostic Laboratory, Iowa State University) for technical advice in the design of the real-time PCR primers and probes, Dr. Kenneth Platt (College of Veterinary Medicine, Iowa State University) for providing the wild-type PRV, Dr. Sabrina Swenson (National Veterinary Services Laboratory, USDA) for providing tissue samples from PRV field cases, and the staff of the virology section at the Iowa State Veterinary Diagnostic Laboratory for their assistance in sample collection and laboratory testing. Authors would also like to thank the following biological companies: Pfizer Animal Health, Schering-Plough Animal Health, Intervet Inc., Boehringer-Ingelheim Vetmedica, and Fort Dodge Animal Health for providing the gE-deleted marker vaccines. This project was supported in part by funding from a USDA-ARS Specific Cooperative Agreement (58–6325–2–0146) and USDA National Animal Health Laboratory Network Cooperative Agreement for CSF surveillance testing.
Footnotes
a.
American Tissue Culture Collection, Manassas, VA
b.
Invitrogen Corp., Carlsbad, CA.
c.
Applied Biosystems, Foster City, CA.
d.
Qiagen Inc., Valencia, CA.
e.
Thermo Fisher Scientific, Waltham, MA.
f.
Cepheid Inc., Sunnyvale, CA.
g.
Promega Corp., Madison, WI.
h.
VMRD Inc., Pullman, WA.