
Abstract
Swine pasivirus 1 (SPaV-1) was first detected in the feces of healthy pigs in France as a new species in family Picornaviridae. We investigated the presence, distribution, and genetic variability of this virus in 7 geographic areas with intensive pig breeding farms in eastern Romania. A total of 564 porcine specimens, including 82 fecal specimens and 482 pools of organs, were collected from healthy pigs in different stages of production from pathogen-free swine farming units. The virus was found in 6 of 7 areas investigated. Of the 564 samples analyzed, 218 were positive for SPaV-1, with the highest prevalence of the virus in organ homogenates (39% positive) followed by feces (37% positive). The highest susceptibility to infection was found in nurseries (50% positive in both the first and second months of feeding). Sequencing analysis of VP0 revealed 3 different Romanian sequences. The phylogenetic investigations suggest that the Romanian sequences cluster with other Pasivirus strains selected from the GenBank database, forming a separate clade from other Picornaviridae genera and defining the described Pasivirus.
Picornaviridae is a large family of small, nonenveloped, icosahedral viruses with positive single-strand RNA genomes. 8 In 2014, the International Committee on Taxonomy of Viruses (ICTV) recognized the division of Picornaviridae family into 29 genera. Some features, such as some conserved regions of the viral genomes, motifs, or cleavage sites corresponding to capsid protein and the nonstructural protein 2C, 3Cpro, and 3Dpol are shared by different genera of picornaviruses. The sequences of other nonstructural proteins (leader [L] protein, 2A, 2B, 3A, and 3B) are not conserved and may have characteristics unique to some novel Picornavirus genera.5,10 For example, the novel genus Pasivirus 13 was proven to lack the L protein, conserved H-box and NC-box motifs, and a putative transmembrane domain present in the 2A protein of parechoviruses and duck hepatitis A virus. 7
Picornaviruses cause a wide range of diseases, have a large variety of hosts, and some are zoonotic (Human enterovirus B [swine vesicular disease virus], Foot-and-mouth disease virus, encephalomyocarditis virus). 11 As a result of the falling cost of nucleic acid sequencing during recent years, many novel picornaviruses have been discovered and characterized,2,5,7,9,10 but it is important to understand the potential for cross-species transmission of these new viruses found in animal hosts. 16 This is the case of swine pasivirus 1 (SPaV-1; Pasivirus A), which was first detected in France in the feces of healthy pigs. 13 SPaV-1 is a new species in the family Picornaviridae and, because is most closely related to Parechovirus B (Ljungan virus), it was placed in a new Parecho sister clade (Pasivirus). SPaV-1 infection was shown to be asymptomatic in piglets, but few studies have been performed regarding SPaV-1 epidemiology in Europe. 13 PLV-CHN (parechovirus-like virus), a novel virus discovered in China, 17 has 82% nucleotide (nt) identity (89% aa) with the French SPaV-1 and is considered a different genotype of Pasivirus.
Eastern Romania is an important region for animal husbandry without any strict surveillance measures, especially for newly emerging diseases. Therefore, we investigated the presence, distribution, and genetic variability of SPaV-1 in eastern Romania.
A total of 564 porcine specimens, including 82 fecal specimens and 482 organ homogenates (tonsil, mediastinal lymph node, spleen, kidney), were collected in 2013–2014 from asymptomatic piglets and adult pigs in different production stages from 24 intensive pig breeding farms from 7 geographic areas (Iasi, Neamt, Buzau, Braila, Vrancea, Ialomita, and Constanta counties; Fig. 1). All samples were deposited in RNA buffer (Ambion RNAlater, Qiagen, Valencia, CA) for transportation and then stored at −80°C.
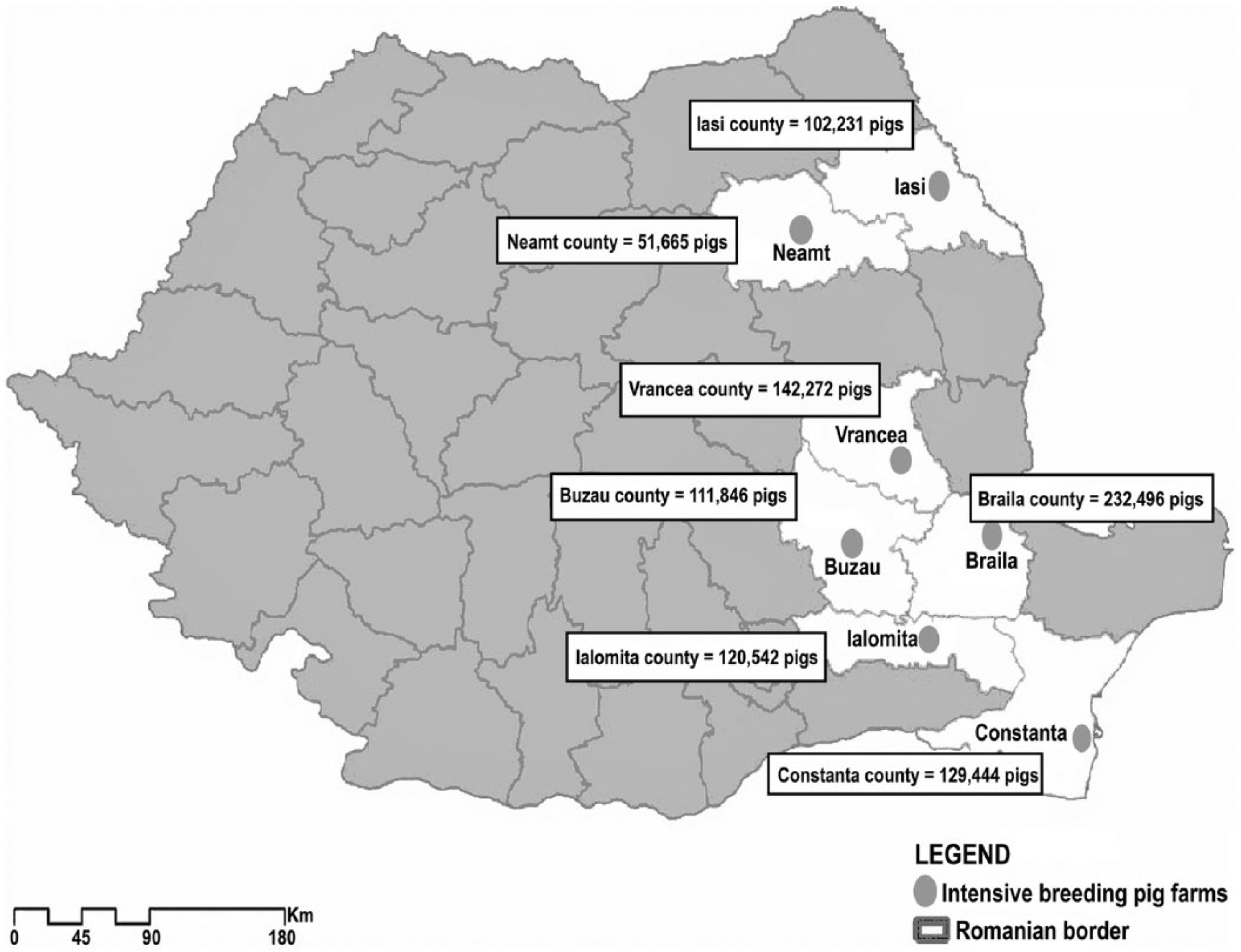
Location of the pig farms investigated in eastern Romania.
Fecal samples were diluted (0.1 g/mL) in phosphate-buffered saline (Gibco, Life Technologies, Grand Island, NY), well homogenized, and then centrifuged at 12,000 × g for 30 min at 4°C. The supernatants were microfiltered through 0.45-µm pore size membranes (Sartorius, Gottingen, Germany) to remove residual eukaryotic and bacterial cell particles.
Total RNA was extracted using an RNA isolation kit (RNeasy mini-kit, Qiagen). A volume of 200 μL of tissue homogenate or fecal supernatant was processed in accordance with the manufacturer’s instructions. The purity and integrity of RNA were evaluated using a nanospectrophotometer (BioSpec–nano, Shimadzu Biotech, Kyoto, Japan).
The virus was detected (GoTaq 1-step RT-qPCR system, Promega, Madison, WI) by obtaining an amplified fragment of 151 bp within the 3D genomic region encoding the RNA-dependent polymerase by using PCR primers (SPaV1.3D.151F: 5′-AAACCATGGCCTGGTGTGCGT-3′; SPaV1.3D.151R: 5′-TGCCAATCGCAGAGTCAACCT-3′), as described previously. 13 The positive control was feces collected from French piglets shedding SPaV-1, 7 diluted (0.1 g/mL) in phosphate-buffered saline (Life Technologies).
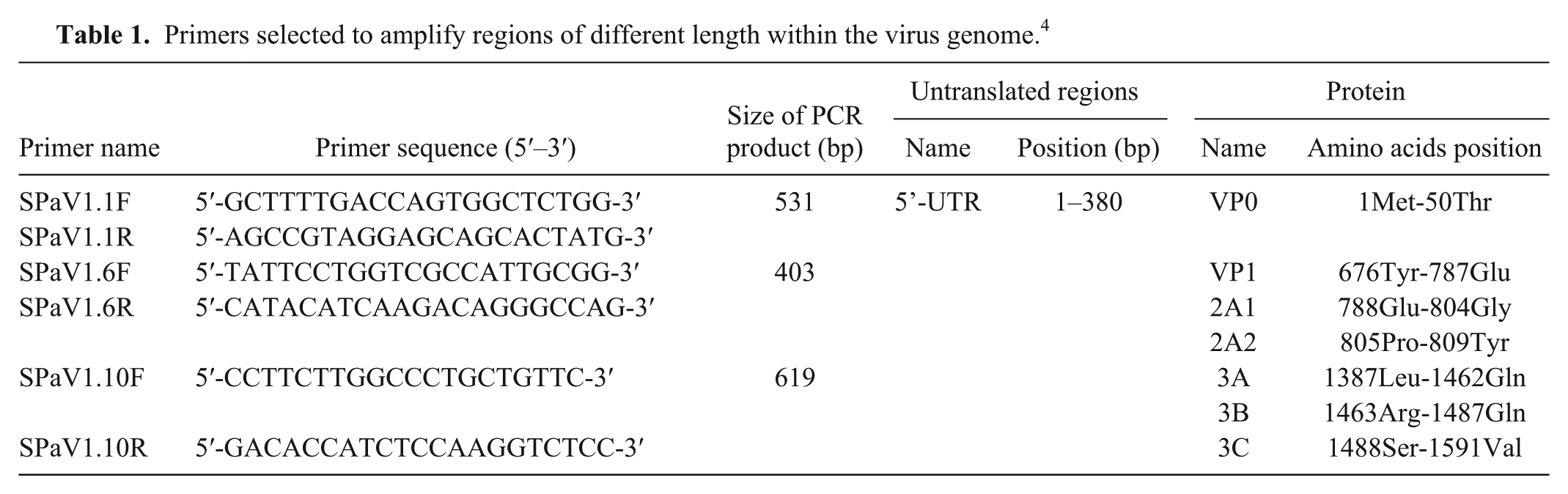
The reaction (Promega) was carried out in a final volume of 20 μL and a final concentration of 0.6 μM for each primer. Polymerase chain reaction (PCR) was performed (LightCycler 2.0, Roche Diagnostics, Rotkreuz, Switzerland) with the following program: 15 min at 42°C, 1 cycle; 10 min at 95°C, 1 cycle; 10 s at 95°C, 30 s at 60°C, 30 s at 72°C, 45 cycles; and 30 s at 40°C, 1 cycle. All 218 positive samples detected by reverse transcription (RT)-PCR were amplified (OneStep RT-PCR kit, Qiagen, Hilden, Germany), and 3 sets of primers (TIB MolBiol, Berlin, Germany) 13 amplified 3 fragments within the viral genome (Table 1). RT-PCR was carried out in a final volume of 25 μL according to the manufacturer’s instructions. The PCR mix contained 0.6 µM forward and reverse primers. PCR was performed (iCycler, Bio-Rad Laboratories, Hercules, CA) with the following program: 30 min at 50°C, 1 cycle; 15 min at 95°C, 1 cycle; 45 s at 94°C, 45 s at 62°C, 60 s at 72°C, 40 cycles; 10 min at 72°C. PCR products were visualized on 1% agarose gels stained with ethidium bromide. The amplified fragments were purified (Wizard PCR Preps DNA purification system, Promega), and DNA sequencing reactions were performed (BigDye Terminator kit v3.1, Applied Biosystems, Foster City, CA; 3130 genetic analyzer, Applied Biosystems).
Primers selected to amplify regions of different length within the virus genome. 4
The 218 samples that were positive for VP0 were sequenced (3130 genetic analyzer, Applied Biosystems). Only 156 high-quality sequences were obtained for VP1/2A and partial 3A/3B/partial 3C regions. The sequenced fragments were edited 6 and deposited in GenBank (accessions KJ780020–KJ780024). The sequence identity matrix of the nucleotide sequences from our study and other SPaV nucleotide sequences (available in GenBank) was performed (MegAlign program, DNASTAR, Madison, WI).
The sequences were aligned using the ClustalW option found in MEGA6. 15 This resulted in a 531-nucleotide alignment for VP0, a 403-nucleotide alignment for VP1/2A, and a 619 nucleotide alignment for partial 3A/3B/partial 3C. The phylogenetic trees were generated in MEGA6 using the neighbor-joining method. 12 The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. 4 The evolutionary distances were computed using the maximum composite likelihood method 14 and are in the units of the number of base substitutions per site (Fig. 1).
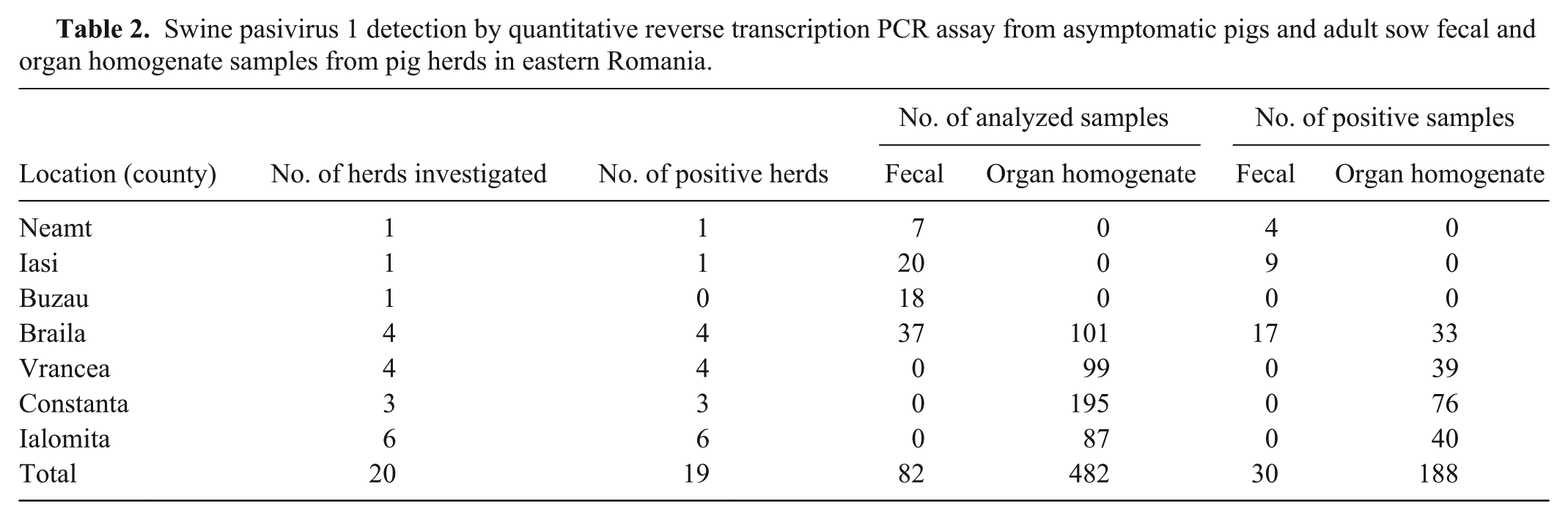
Prevalences of SPaV in fecal samples and tissue homogenates varied among geographic regions (Table 2). Of the total samples, 39% (218 of 564) were positive for SPaV-1 by RT-PCR (Table 3).
Swine pasivirus 1 detection by quantitative reverse transcription PCR assay from asymptomatic pigs and adult sow fecal and organ homogenate samples from pig herds in eastern Romania.
The prevalence of swine pasivirus 1 between virus detection in pigs and production stage.

The territorial distribution analysis of SPaV-1 in eastern Romania revealed the presence of the virus in 6 of 7 investigated areas. The prevalence of SPaV-1 varied with its geographic distribution. Only the farm from Buzau showed no positive samples for SPaV-1 (Table 2). However, given the small number of samples analyzed for Buzau, a conclusion on the presence or absence of infection in this area could not be stated.
In suckling piglets, 40% (33 of 82) of the samples were found positive for SPaV-1, but the highest prevalence of the virus was found in the first and second months of feeding (50%) before decreasing to 25% (3 of 12) in the third month of feeding (Table 3). A much lower rate of virus prevalence was found in sows (11%; 9 of 82). However, given the small sample size from these production stages, further investigation is needed in order to strengthen the conclusion concerning the presence of the virus in pigs in the second and the third months of feeding. The presence of the virus estimated as a percentage from all analyzed samples was similar for the 2 different types of specimens (feces and organ homogenates). The difference appears in the second month of feeding group in which the prevalence of SPaV-1 in pig feces was much lower (22%) than in organ homogenates (63%). This occurrence could be associated with decreased maternal antibodies. Another hypothesis is the possible accumulation of molecular variations in the genomic sequences of the virus that might lead to antigenic differences, a mechanism used by many viruses to avoid the host immune response. 1
The alignment of the sequenced fragments revealed 3 different nucleotide sequences specific to Romania and corresponding to the amplified VP0 genomic region (Rom1, Rom2, and Rom3). Only the Rom1-specific sequence (GenBank accession KJ780020) was found in all investigated areas. The other 2 sequences were each specific to a single region: Rom2 (accession KJ780021) for Iasi, and Rom3 (accession KJ780022) for Vrancea. Analyzing the specific VP1/2A nucleotide alignment, only 1 Romanian-specific nucleotide sequence (accession KJ780023) was detected.
The alignment for the partial 3A/3B/partial 3C sequences also revealed 1 nucleotide sequence specific for eastern Romanian pig farms (GenBank accession KJ780024). Nevertheless, it should be noted that because it was not possible to get PCR products for VP1/2A and partial 3A/3B/partial 3C genomic regions from all samples, variation in genetic sequence within those regions may have escaped detection. Of 218 positive samples, only 156 were sequenced for all 3 genomic regions. The results showed that at least 3 strains of SPaV-1 circulate in eastern Romania and that the highest sequence diversity is likely located in the VP0 sequence.
Sequence identity matrix analysis for Rom1 and Rom2 showed greater sequence similarities to VP0 coding regions belonging to SPaV-1 (0.877 and 0.871, respectively) than to those belonging to PLV-CHN (0.777 and 0.775, respectively). Rom3 was more similar to PLV-CHN (0.795) than to SPaV-1 (0.733). The most similar sequences were found to be Rom1 and Rom2 (0.924). Also, VP1/2A sequences of the Romanian virus showed higher similarity with SPaV-1 than with PLV-CHN. Partial 3A/3B/partial 3C fragment showed significant sequence similarities with both SPaV-1 and PLV-CHN (Table 4).
Nucleotide similarities of the VP0, VP1/2A, and partial 3A/3B/partial 3C regions to the swine pasivirus 1 (SPaV-1) and PLV-CHN strains.*

PLV-CHN = parechovirus-like virus discovered in China; dash (–) = no nucleotide similarities analysis.
SPaV-1 has been included in the same clade as genera Parechovirus, Avihepatovirus, and Aquamavirus. 13 The Chinese isolate PLV-CHN and SPaV-1 cluster tightly together compared to other picornaviruses. 17
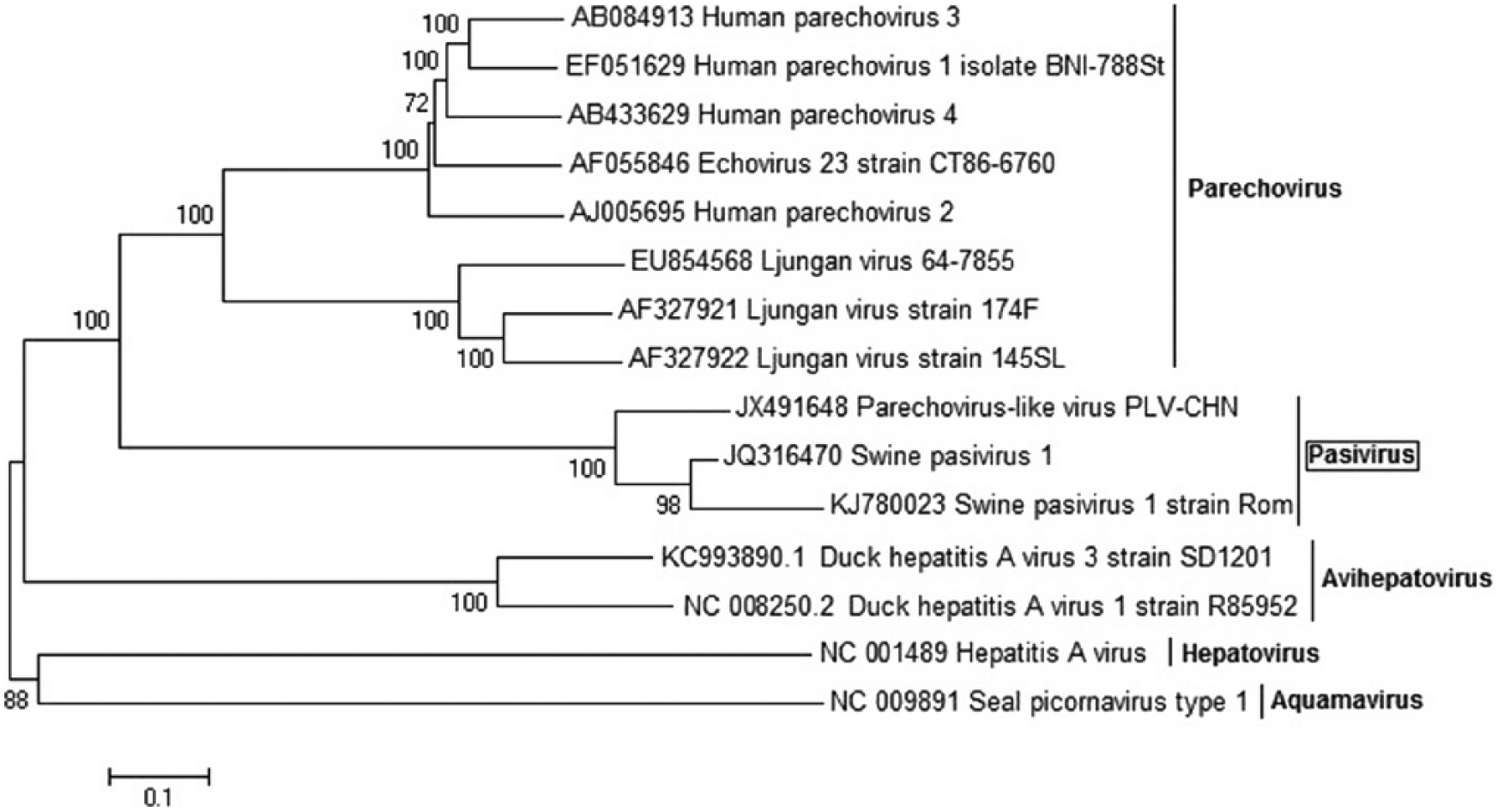
In order to visualize the relationships among the Romanian virus and other isolates, 3 phylogenetic trees were constructed based on the pairs of primers used (SPaV1.1, SPaV1.6, and SPaV1.10). The phylogenetic investigations based on VP0 suggested that Rom1, Rom2, and Rom3 sequences, together with the other Pasivirus strains, form a separate clade of the Picornaviridae genera (Fig. 2). Comparing pairwise sequence identity between Romanian nucleotide sequences, the results showed that Rom1 and Rom2 are the most related (92% identity) and, together, share only 74% identity with Rom3. Pairwise sequence identity analysis revealed that the sequence of nucleotides belonging to the Romanian fragments Rom1 and Rom2 shared 87.7% and 87.1% identity, respectively, with the corresponding VP0 sequence belonging to SPaV-1 and a lower nucleotide identity (77.7% and 77.5%, respectively) when compared with the corresponding VP0 nucleotide sequence belonging to PLV-CHN. However, Rom3 shared a higher sequence identity with the corresponding VP0 fragment belonging to the Chinese isolate (79.5% identity) than with the VP0 sequence of the European strain (73.3% identity).
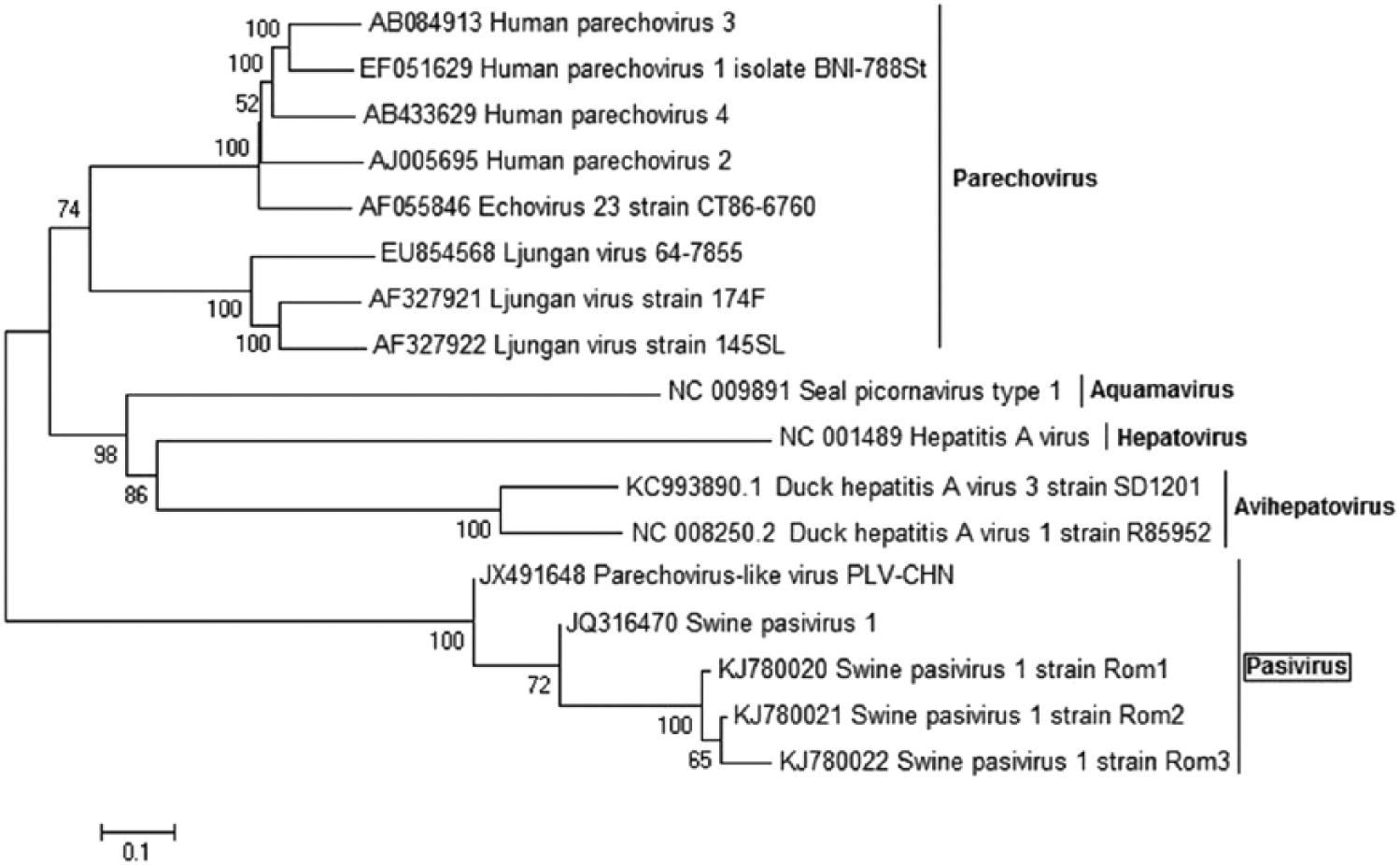
Phylogenetic analysis of the Romanian isolates specific for VP0 nucleotide sequences. The tree was constructed using the neighbor-joining method with 1,000 bootstrap replicates.
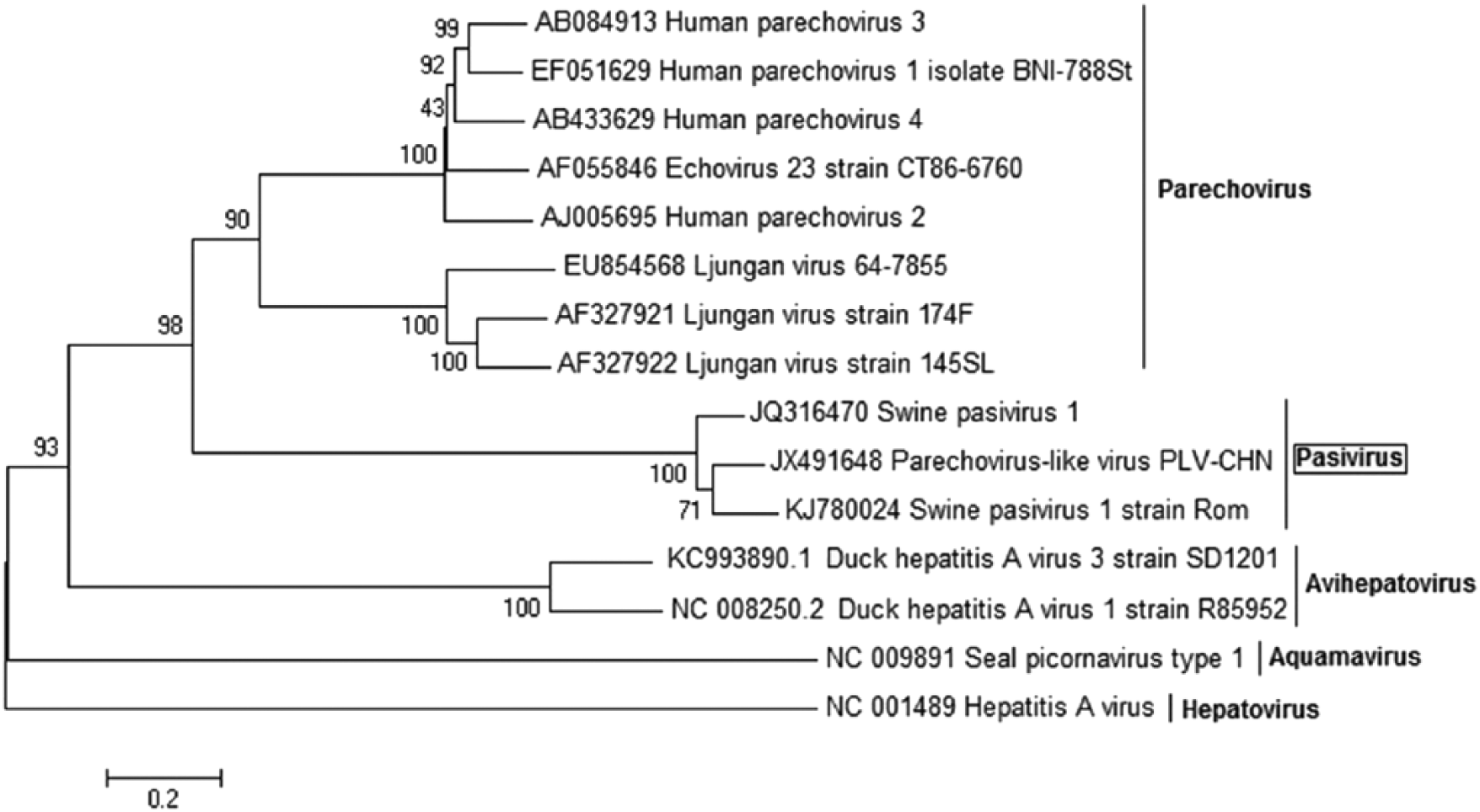
The same topology of the phylogenetic tree was discovered for the VP1/2A region (Fig. 3). In this case, the Romanian sequence forms a distinct clade with the French and Chinese isolates. Pairwise sequence identity analysis revealed that the Romanian VP1/2A sequence is most closely related to the French corresponding VP1/2A fragment (85.3% identity with SPaV-1; 72.4% identity with PLV-CHN).
Phylogenetic analysis of the Romanian isolates specific for VP1/2A nucleotide sequences. The tree was constructed using the neighbor-joining method with 1,000 bootstrap replicates.
The Romanian nucleotide sequence for partial 3A/3B/partial 3C also clusters with SPaV-1 and PLV-CHN (Fig. 4), sharing an identity of 83.5% and 83.1%, respectively, with the corresponding partial 3A/3B/partial 3C fragments belonging to the French and Chinese isolates. Phylogenetic analysis placed the Pasivirus genus in closer relation to members of the genus Parechovirus than to the members of other analyzed genera (Hepatovirus, Aquamavirus, and Avihepatovirus).
Phylogenetic analysis of the Romanian isolate specific for partial 3A/3B/partial 3C nucleotide sequences. The tree was constructed using the neighbor-joining method with 1,000 bootstrap replicates.
The high prevalence of SPaV-1 in almost all pig farms from eastern Romania investigated during our study and also the high prevalence among the investigated pigs within each farm should be reasons for continuing the investigation of this genus. Little is known about SPaV-1, and there is no information regarding the mechanics of its pathogenicity. The infected pigs do not display any signs of infection. The manner of distribution of SPaV-1 in pigs in different production stages leads to the conclusion that the virus is shed continuously and that it disseminates in the host most likely by viremia.3,13
Footnotes
Acknowledgements
We thank Drs. Gheorghe Savuta and Adriana Anita from the Faculty of Veterinary Medicine, University of Agricultural Sciences and Veterinary Medicine, Iasi.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Project QualiMeat–PN-II-PT-PCCA-2011-3.2-0509 and by the strategic grant POSDRU/159/1.5/S/133391, Project “Doctoral and Post-doctoral programs of excellence for highly qualified human resources training for research in the field of Life sciences, Environment and Earth Science” co-financed by the European Social Fund within the Sectorial Operational Program Human Resources Development 2007–2013.