
Abstract
Capripoxviruses (CaPVs), consisting of Sheeppox virus (SPV), Goatpox virus (GPV), and Lumpy skin disease virus (LSDV) species, cause economically significant diseases in sheep, goats, and cattle, respectively. Quantitative real-time polymerase chain reaction (qPCR) assays are routinely used for rapid detection of CaPVs in surveillance and outbreak management programs. We further modified and optimized 2 previously published CaPV qPCR assays, referred to as the Balinsky and Bowden assays, by changing commercial PCR reagents used in the tests. The modified assays displayed 100% analytical specificity and showed no apparent changes in analytical sensitivities for detection of CaPVs compared with the original assays. Diagnostic sensitivities, assessed using 50 clinical reference samples from experimentally infected sheep, goats, and cattle, improved from 82% to 92% for the modified Balinsky assay and from 58% to 82% for the modified Bowden assay. The modified qPCR assays were multiplexed for detection of beta-actin as an indicator for potential false-negative results. The multiplex modified qPCR assays exhibited the same diagnostic sensitivities as the singleplex assays suggesting their utility in the detection of CaPVs.
Sheeppox virus (SPV), Goatpox virus (GPV), and Lumpy skin disease virus (LSDV) are species in the genus Capripoxvirus (CaPV) within the family Poxviridae, subfamily Chordopoxvirinae.5,15 Sheep, goats, and cattle are susceptible to infection by SPV, GPV, and LSDV, respectively. These viruses are considered reportable agents to the World Organization for Animal Health (OIE) given their potential for significant economic impact. Capripox is endemic in many parts of the world including most of Asia (SPV and GPV), Africa (SPV, GPV, and/or LSDV), and the Middle East (SPV, GPV, and LSDV). 1 CaPVs are closely related, with genomic sequence identities ranging from 96% between the species to 99% between virus strains of the same species. 15 Molecular assays have been reported for rapid detection of CaPVs, including quantitative real-time polymerase chain reaction (qPCR)2 –4,11 and loop-mediated isothermal amplification.6,12
We report herein further modification and improvement of 2 previously published CaPV qPCR assays, the Balinsky assay 3 and the Bowden assay. 4 The Balinsky assay targets the poly (A) polymerase small subunit gene (ORF068), and the Bowden assay targets the intracellular mature virion envelope protein gene (ORF074) along with the host 18S ribosomal RNA gene (as an endogenous control) or a commercial plasmid (as an exogenous control). The target amplicons for both assays are well conserved within the CaPV genus and were derived from the LSDV Neethling 2490 genome sequence (GenBank accession AF325528.1). The original multiplex Bowden assay was developed on the ABI 7700 thermocycler, and its performance has not been tested on the SmartCycler, a commonly used thermocycler in many veterinary diagnostic labs in the United States. In contrast, the original singleplex Balinsky assay was developed for the SmartCycler; however, the PCR kit (Kit-EZ; GeneAmp EZ rTth RNA PCR, Life Technologies, Carlsbad, CA) used in the original assay has been discontinued by the manufacturer. We report further modification of both assays by changing the thermocycler for the Bowden assay and by changing the PCR kits for both assays. In addition, both assays were multiplexed to detect the beta-actin gene (ACTB) as an endogenous positive control to detect PCR inhibition (false-negatives) in diagnostic specimens.
The CaPV isolates SPV-HELD, SPV-6LT-1, GPV-Pendik, GPV-Turkey, LSDV-Makhana, LSDV-Uganda, LSDV-Ismalia, and LSDV-Cameroon were obtained from the Reagents and Vaccines Services Section of the Foreign Animal Disease Diagnostic Laboratory, Plum Island Animal Disease Center (Greenport, NY). The CaPV isolates SPV-Nigeria, GPV-India, and LSDV-Neethling were obtained as infected clinical specimens (Canadian Food Inspection Agency, Winnipeg, Canada). The clinical specimens used in our study included swabs (oral, nasal, and conjunctival), EDTA blood, and tissues (skin lesions, skin nodules, scabs, and lung) from sheep, goats, and cattle experimentally infected with SPV-Nigeria, GPV-India, and LSDV-Neethling, respectively, as described earlier.2,4,6 Negative specimens, including swabs, EDTA blood, and skin were collected from normal healthy cattle and were provided as generous gifts from the University of Georgia Athens Veterinary Diagnostic Laboratory, Athens, Georgia. The CaPV isolates were grown on primary lamb testis cells as described previously. 6
Tissue samples were washed in 1× phosphate-buffered saline (Life Technologies, Carlsbad, CA) and suspended in Dulbecco modified Eagle medium (Life Technologies) at a final concentration of 10% (w/v) and homogenized in grinding jars (tissue lyser, Qiagen, Valencia, CA) for 2 min at an oscillation frequency of 22 Hz. Tissue homogenates or swabs at 200 µL each, or 100 µL EDTA blood samples, were used as starting material for DNA extraction using an extraction kit (DNeasy blood and tissue kit, Qiagen) according to the manufacturer’s instructions. Extracted DNA was eluted in 100 µL of kit-supplied elution buffer (buffer AE).
The original Balinsky assay3,6 was performed using Kit-EZ and the original Bowden assay2,4 was performed using Kit-TU (TaqMan universal PCR master mix, Life Technologies). Both assays were performed on the SmartCycler (SmartCycler II, Cepheid, Sunnyvale, CA) using 2.5 µL of template DNA in a final reaction volume of 25 µL. The PCR primers (Integrated DNA Technologies, Coralville, IA) and the fluorogenic hydrolysis probes (Life Technologies) used for both assays were obtained from commercial sources. The probes were labeled with FAM (6-carboxyfluorescein) at the 5′-end and minor groove binder (MGB) nonfluorescent quencher (MGBNFQ) at the 3′-end. Two new commercial PCR kits, Kit-PD (Path-ID multiplex one-step RT-PCR kit, Life Technologies) and Kit-TFV, (TaqMan Fast virus 1-step master mix, Life Technologies) were evaluated for use with the qPCR assays as a modification. The performance of the modified assays using the new PCR kits was tested using serial dilutions of DNA extracted from cell culture–grown CaPVs (SPV-HELD, GPV-Pendik, and LSDV-Makhana) in buffer TE (10 mM Tris–1 mM EDTA, pH 8.0) (Integrated DNA Technologies) as template. Optimum sensitivity of detection of the viral DNA was obtained using Kit-TFV for both assays. Therefore, all modified qPCR assays reported in our study were performed using Kit-TFV. The composition of reaction master mixes of the modified assays included the same concentrations of CaPV-specific primers and probe as used in the original assays,3,4 plus 6.25 µL of the kit-supplied 4× master mix of Kit-TFV, 2.5 µL of template DNA, and the balance of nuclease-free water in a final reaction volume of 25 µL. Thermocycling conditions for the qPCR assays performed with Kit-TFV included 1 cycle of heat denaturation at 95°C for 20 s followed by 40 cycles of 95°C for 3 s, and 60°C for 30 s. All qPCR assays were performed using a minimum of 3 replicates along with appropriate controls, including a positive amplification control (SPV-HELD DNA) and a no-template control TE. No difference in the threshold cycle (Ct) values was found using either nuclease-free water or TE as a diluent; therefore, all serial dilutions of the templates used in our study were prepared in TE.
Recombinant positive control (PC) plasmids were used as reference standards to determine the analytical sensitivity (ASe) or limit of detection (LOD) of the CaPV qPCR assays. The PC plasmid used for the Balinsky assay was pORF068, as described previously. 6 The PC plasmid used for the Bowden assay, referred to as pORF074, was constructed by cloning PCR-amplified, full-length ORF074 (969 bp) of LSDV (GenBank accession AF325528.1) into a plasmid vector using a commercial cloning kit (TOPO TA cloning kit, Life Technologies). The PCR product (969 bp) was amplified from LSDV (Makhana) DNA with a PCR kit (AmpliTaq Gold 360 master mix, Life Technologies) using 500 nM each of the forward (5′-ATGGCAGATATCCCATTATA-3′) and reverse (5′-CTAAACTATATACGTAAATAACATACC-3′) primers (Integrated DNA Technologies) under thermocycling conditions described previously. 6 Plasmids pORF068 and pORF074 were transformed and propagated in Escherichia coli competent cells (Mach1-T1R Escherichia coli competent cells, Life Technologies) and extracted (PureLink HiPure mini plasmid DNA purification kit, Invitrogen, Carlsbad, CA).
Quantified pORF068 and pORF074 were serially diluted in TE and used as template to determine the LOD of the modified Balinsky and Bowden assays, respectively. A standard curve was prepared by plotting the Ct values against the copy number of the PC plasmid to determine the LOD, expressed as copy number of the plasmid per assay where each copy of the plasmid represents 1 genome copy of the virus particle. Plasmid copy number was calculated using the formula: copy number = [mass (g) × 6.022 × 1023]/[length (bp) × 650 daltons].
The modified qPCR assays were multiplexed to detect ACTB as an internal positive control. The forward (5′-TGACATCAAGGAGAAGCTCTGC-3′) and reverse (5′-CCGCGGTGGCCATCT-3′) primers and the probe (5′-VIC-ACGTGGCCCTGGACTTCGAGCA-TAMRA-3′) sequences were designed from a highly conserved 64-bp region of ACTB from multiple animal hosts including cow, sheep, goat, rabbit, swine, and horse (Fig. 1). Optimal concentrations of the primers (Integrated DNA Technologies) (forward and reverse) and probe (Life Technologies) for amplification of ACTB were determined to be 200 nM each, which were added to master mixes of the modified singleplex Balinsky and Bowden assays. Thermocycling conditions for the multiplex assays were the same as used for the singleplex assays, and the amplification of CaPV and ACTB were monitored in the FAM and cyanine (Cy)3 channels, respectively.
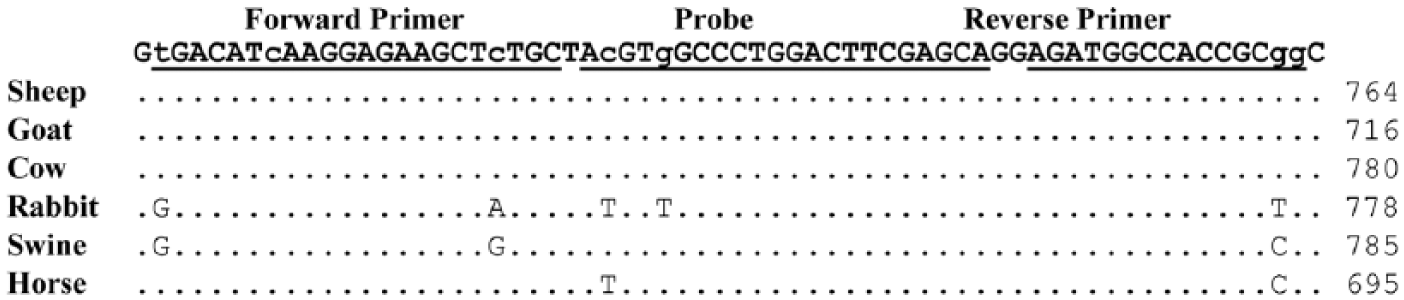
Alignment of the nucleotide
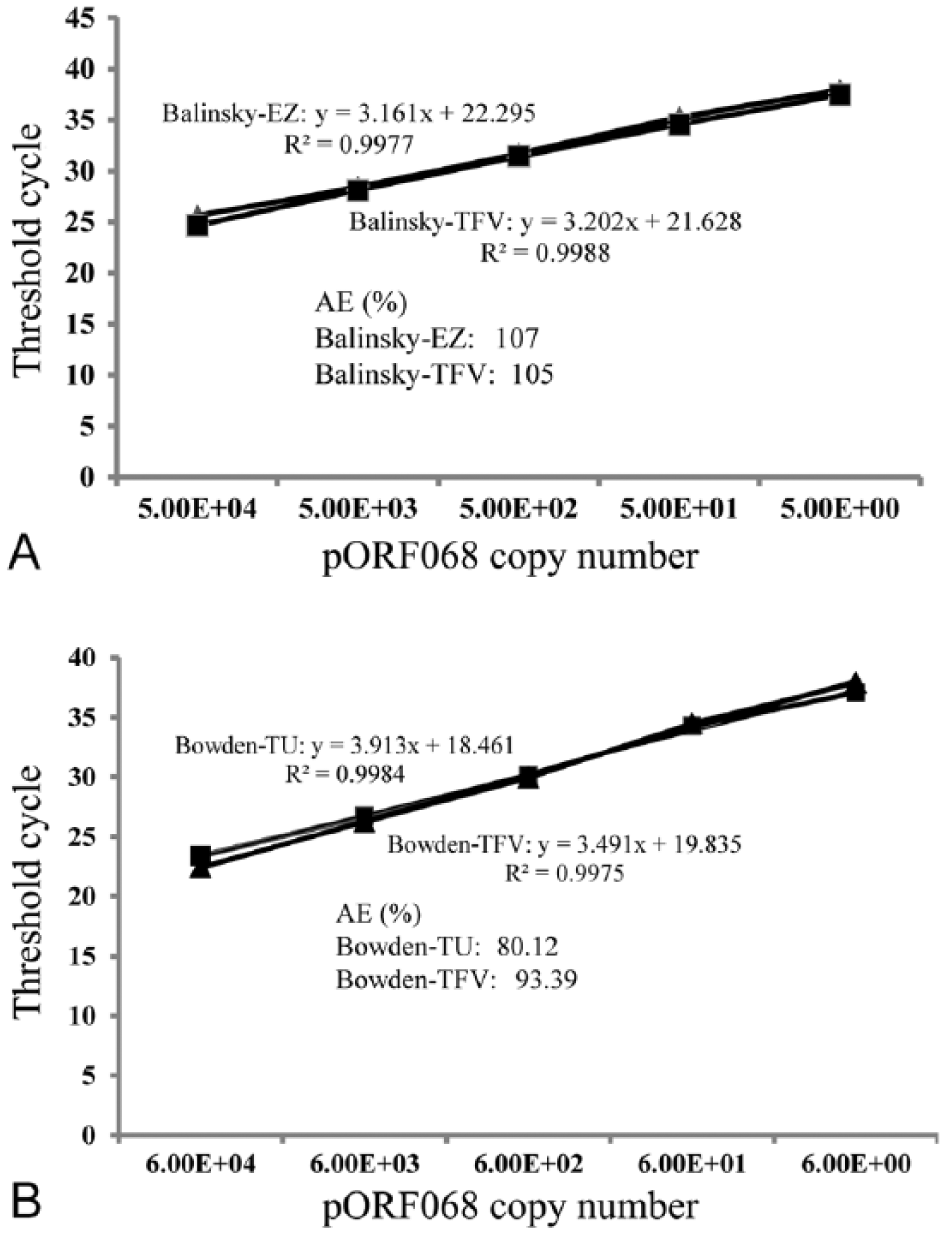
ASe for the qPCR assays, determined against serial dilutions of the recombinant PC plasmid targets mentioned above, was 5 genome copies per assay for both original and the modified Balinsky assays, and 6 genome copies per assay for both original and modified Bowden assays (Fig. 2). These LODs were also in close agreement with those reported previously for both original assays.4,6
ASe was also determined using the DNA extracted from serial dilutions of titered cell culture–grown SPV-HELD, GPV-Pendik, and LSDV-Cameroon. Once again, no change in analytical LODs against virus isolates was observed between the original and the modified assays as performed on the SmartCycler platform (Table 1). The amplification efficiencies calculated from the Ct values (not shown) corresponding to the serial dilutions of the viral DNA was estimated to be 99–110% for all assays.
Limits of detection and amplification efficiencies of capripoxvirus qPCR assays.*
Cell culture–grown SPPV-HELD, GTPV-Pendik, and LSDV-Cameroon were serially diluted in Dulbecco modified Eagle medium. DNA extracted from each dilution in triplicate was used as template. The lowest titer (TCID50/mL) of diluted virus to be detected in triplicate is shown. Amplification efficiency (AE) was calculated from the slope of the standard curve using the formula AE = (10(−1/slope) − 1) × 100. ND = not determined.
Superscript letters refer to the Sources and Manufacturers list.
Sources: Kit-EZ = GeneAmp EZ rTth RNA PCR kit, Life Technologies; Kit-TFV = TaqMan Fast virus 1-step master mix, Life Technologies; Kit-TU: TaqMan universal PCR master mix, Life Technologies.
Analytical specificities of the modified Balinsky and Bowden assays were determined using an inclusivity panel consisting of DNA extracted from multiple cell culture–grown isolates of CaPV species, including SPV (SPV-HELD and SPV-6LT-1), GPV (GPV-Pendik, and GPV-Turkey), and LSDV (LSDV-Makhana, LSDV-Uganda, LSDV-Ismalia, and LSDV-Cameroon). All CaPV isolates of the inclusivity panel tested positive, and there was no cross-reactivity found using an exclusivity panel consisting of other poxvirus species, including Orf virus, Bovine papular stomatitis virus, Pseudocowpox virus, and Swinepox virus.
Diagnostic sensitivities (DSe) were assessed on 50 clinical specimens including swabs (n = 20), EDTA blood (n = 13), and tissues (n = 17) from experimentally infected cattle, goats, and sheep. The number of specimens tested positive were 41 (82%) and 46 (92%) by the Balinsky assay performed with Kit-EZ and Kit-TFV, respectively, and 29 (58%) and 41 (82%) by the Bowden assay performed with Kit-TU and Kit-TFV, respectively (Table 2). All 50 specimens were previously analyzed by loop-mediated isothermal amplification (LAMP) assay, 6 and the 45 positive samples by LAMP (not shown) were all included among the 46 samples that tested positive by the modified Balinsky assay (Table 2).
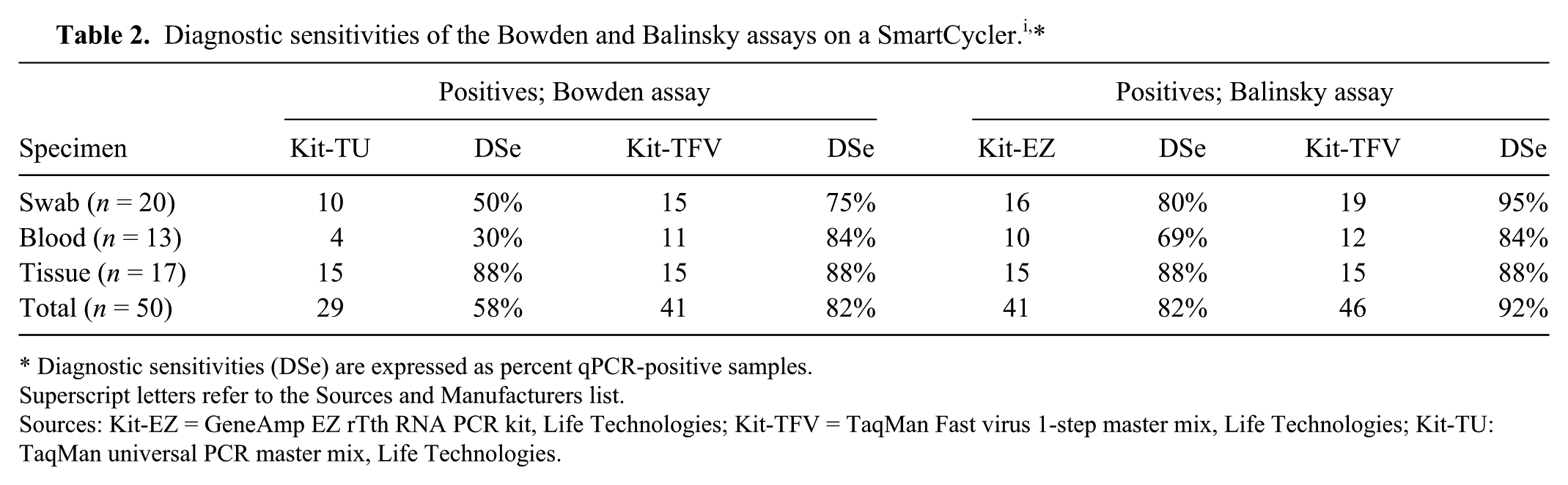
Diagnostic sensitivities (DSe) are expressed as percent qPCR-positive samples.
Superscript letters refer to the Sources and Manufacturers list.
Sources: Kit-EZ = GeneAmp EZ rTth RNA PCR kit, Life Technologies; Kit-TFV = TaqMan Fast virus 1-step master mix, Life Technologies; Kit-TU: TaqMan universal PCR master mix, Life Technologies.
Diagnostic specificities were assessed on 43 negative clinical specimens, including swabs, EDTA blood, and tissues from normal healthy cattle, goats, and sheep. All tested negative (100%) by the modified Balinsky and Bowden assays (results not shown).
Poor sample quality or the presence of naturally occurring PCR inhibitors are the leading causes of false-negative results and poor DSe as reported by others.7–10,13 Multiplexed detection of a housekeeping host gene can aid in identification of false-negative results. In this regard, ACTB, encoding the well-conserved cytoskeletal protein beta-actin, is commonly used as an internal positive control to detect PCR inhibitors in diagnostic specimens.10,14
The modified Balinsky and Bowden assays were further supplemented by addition of primers and probe to enable multiplexed detection of ACTB. To determine if there was any loss of sensitivity because of multiplexing, the modified singleplex (CaPV only) and multiplex (CaPV and ACTB) qPCR assays were performed using LSDV DNA (LSDV-Cameroon) serially diluted in a background of purified DNA extracted from EDTA blood of normal healthy cattle. LOD of the LSDV DNA was determined to be the same (10−5 dilution) for both singleplex and multiplex Balinsky and Bowden assays (results not shown), indicating no loss of sensitivity in the detection of the virus as a result of multiplexing.
DSe of the multiplex Balinsky and Bowden assays was determined using the same positive cohort of 50 clinical specimens as used in the singleplex assays (Table 2), and the results (Table 3) show that the number of specimens that tested positive remained the same as the singleplex assays (Table 2). In addition, the detection of ACTB in all specimens (Table 3), suggests no apparent PCR inhibition. As shown in Table 2, fewer specimens tested positive by the original singleplex Balinsky (Kit-EZ) and Bowden (Kit-TU) assays. These individual samples that tested negative using the original singleplex assays are further indicated in Table 3.
Target detection by capripoxvirus (CaPV) and ACTB quantitative real-time PCR (qPCR) multiplex Balinsky and Bowden assays.*
Ct = threshold cycle; dpi = day post inoculation; Skin-N = skin nodule; Swab-C = conjunctival swab; Swab-N = nasal swab; Swab-O = oral swab.
Tested negative by Balinsky assay with Kit-EZ (GeneAmp EZ rTth RNA PCR kit, Life Technologies).
Tested negative by Bowden assay with Kit-TU (TaqMan universal PCR master mix, Life Technologies) on a SmartCycler II (Cepheid).
Our study describes observed changes in performance characteristics after modification of 2 previously published highly sensitive CaPV qPCR assays. PCR reagents were changed for the Balinsky and Bowden assays, with the Bowden assay also being further adapted to the SmartCycler platform in a singleplex format. Both assays were multiplexed to include detection for ACTB to serve as an internal positive control and indicator of potential false-negative results when testing clinical diagnostic samples or in vitro culture isolates. In this regard, ACTB, as a host gene target, would not be expected to serve as a relevant control when testing environmental samples that may lack sufficient levels of animal DNA.
By modification of the CaPV qPCR assays with or without multiplex detection of ACTB, it was apparent that there was a marginal-to-significant improvement in the detection of CaPVs (+10% DSe for the Balinsky assay and +24% DSe for the Bowden assay) when using Kit-TFV as compared to using the original kits (Kit-EZ or Kit-TU for the Balinsky and Bowden assays, respectively; Tables 2, 3). In this regard, it seems likely that the PCR inhibitors in clinical diagnostic specimens are largely responsible for lesser detection of CaPVs by the Bowden assay performed with Kit-TU (Supplemental Table 1, available online at http://vdi.sagepub.com/content/by/supplemental-data). Based on these findings, we anticipate that the modifications presented will further expand and enhance the utility of the Balinsky and Bowden assays for detection and diagnosis of CaPV diseases in livestock.
Footnotes
Acknowledgements
We thank Drs. Timothy Bowden and David Boyle from the Commonwealth Scientific and Industrial Research Organization (CSIRO) Australia for help in generation of the samples, and Michael Pushka from Canadian Food Inspection Agency, National Centre for Foreign Animal Diseases, Winnipeg, Manitoba, Canada, for providing support for shipment of the experimental samples used in this study.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the USDA, Animal and Plant Health Inspection Service, National Veterinary Services Laboratories as part of the animal disease diagnostic mission and by interagency agreement with the USDA Agriculture Research Service under award number 60-1940-7-011.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.