
Abstract
Two novel 1-step real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) assays for the simultaneous detection of West Nile virus (WNV) lineage 1 and 2 strains were developed. Primers and the probe of assay 1 target the 5′-untranslated region (UTR), whereas the amplicon of assay 2 is located in the nonstructural region NS2A, which enables an unambiguous and independent WNV diagnosis based on 2 different amplicons. Both assays allow the detection of as few as 2–4 genome copies of WNV strains NY99, Uganda B956, Kunjin, and Sarafend (all cultured on Vero cells). A new synthetic RNA mutant of the 5′-UTR amplicon, which contains 6 twist inverted base-pair changes at the probe attachment site, was used as external calibrator control.
West Nile virus (WNV; family Flaviviridae, genus Flavivirus) was first detected in a woman in the West Nile District of Uganda in 1937. 24 West Nile virus is an arthropod-borne virus and grouped in the Japanese encephalitis virus (JEV) serocomplex, which includes the St. Louis encephalitis virus, JEV, and Murray Valley encephalitis virus (MVEV), among others. 11 A large variety of wild bird species are the natural reservoir for WNV 19 ; however, its host range is very broad and encompasses not only humans but also equids, alligators, dogs, sheep, and many other species. 4 Human infections are characterized by flu-like illnesses that are associated with headache, high fever, chills, arthralgia, malaise, and retro-orbital pain. Up to 1% of infected human beings develop severe encephalitis, myelitis, and/or meningitis, and of these cases, 1 in 20 dies. 20
The introduction of WNV into New York in 1999 and its rapid spread lead to cases in almost all North American states and provinces, in addition to some Central and South American countries. 10 In Europe, WNV was first detected in France 14 and Portugal, 9 and recent outbreaks have occurred in Romania, 21 Italy, 18,22 Hungary, 8 and Austria. 25
West Nile virus consists of a linear, single-stranded, plus-sense RNA, which encodes for 3 structural (C, prM, and E) and 7 nonstructural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) proteins. 4 It has been recently proposed 3 that WNV be grouped into 5 lineages. Lineage 1 is found in some parts of southern Europe, Asia, Africa, and North America. The Kunjin virus, which circulates in Australia, represents a subtype of lineage 1. 23 Lineage 2 strains are found in sub-Saharan Africa and Madagascar 5 and have also recently been discovered in Hungary and Austria. 2 Lineage 3 is represented by a virus strain that was isolated from mosquitoes in the Czech Republic, designated the Rabensburg virus1; lineage 4 was isolated from a tick isolate from the Caucasus. 2 West Nile virus strains from India, which group into a subcluster of lineage 1, are sometimes thought to represent lineage 5. 3
The aim of the following study was to develop 2 one-step duplex real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) assays that target different regions of the WNV genome for an unambiguous identification of viral nucleic acid. For this purpose, the WNV strains NY99 a (GenBank accession no. AF196835), Uganda B956 a (GenBank accession no. AY532665), Sarafend b (GenBank accession no. AY688948), and Kunjin b (GenBank accession no. D00246) were used, which were grown on Vero (African green monkey kidney epithelial) cells. Virus titers were determined by 10-fold dilution series (in 8 replicates) in 96-well plates (100 μl/well) on fresh cell monolayers and cytopathologic effects, read after an incubation period of 6–7 days at 37°C. Cells were subsequently fixed and stained with crystal violet. Virus titers (50% tissue culture infective doses [TCID50]) were calculated using the Spearman–Kärber method. 15 Moreover, Yellow fever virus (YFV) strain YF-17D, c JEV, d Tickborne encephalitis virus (TBEV) strain Langaat, e and MVEV b were used in the qRT-PCR specificity studies. Viral RNA was isolated from a cell culture medium using a commercial kit. e Cell culture supernatant (140 μl) was added to 560 μl AVL (lysis) buffer, e spiked with 5 μl of an internal control RNA (IC-RNA) containing 2 × 10 5 copies/μl, and eluted from columns in a final volume of 50 μl in AVE buffer e and stored at −70°C until use.
Suitable primers and probes for the qRT-PCR detection of WNV lineage 1 and 2 strains were designed in silico by aligning full-length sequences of 186 flavivirus and 95 WNV isolates (from the National Center for Biotechnology Information database) using Vector NTI Advance primer design software 10.0. f The first WNV-specific amplicon site was identified in the highly conserved 5′-untranslated region (UTR) segment (assay 1) and the second in the nonstructural NS2A region (assay 2). The corresponding primers and probes are listed in Table 1. Probes were labeled at the 5′ end with the FAM reporter dye and at the 3′ end with the quencher dye TAMRA. g
Primers and probes selected for West Nile virus (WNV)–specific real-time quantitative reverse transcription polymerase chain reaction. *
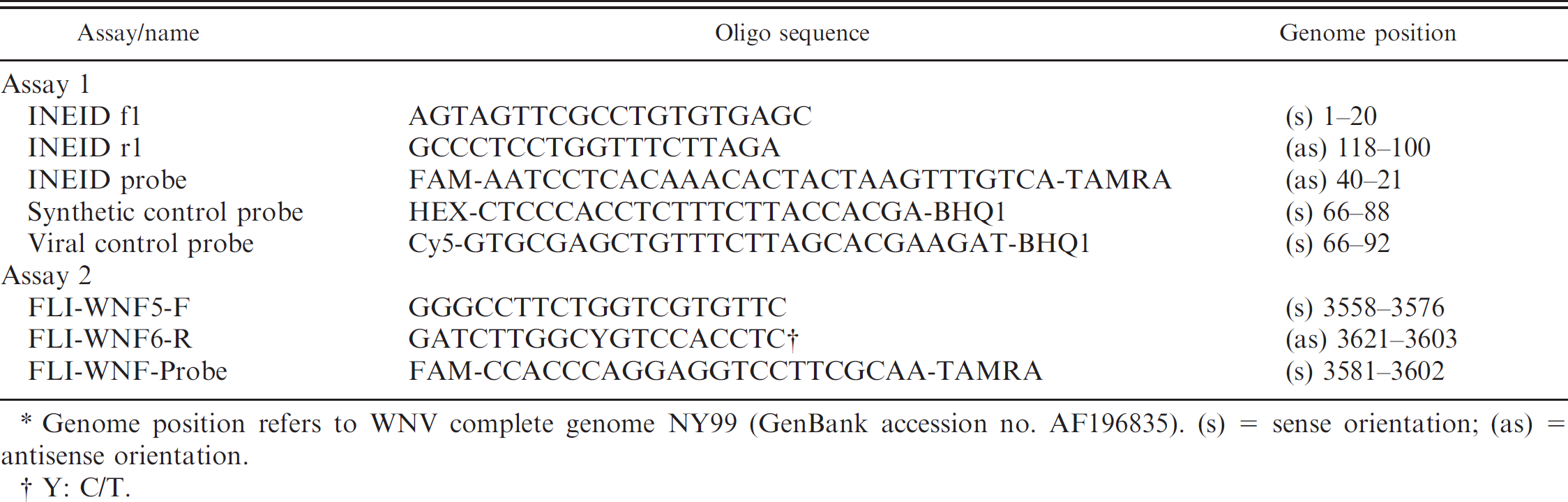
Genome position refers to WNV complete genome NY99 (GenBank accession no. AF196835). (s) = sense orientation; (as) = antisense orientation.
Y: C/T.
The real-time PCR assays were performed with a commercial system h and kit e in a total volume of 25 μl. For these assays, 5 μl of RNA, 20.0 pmol of each primer, and 2.5 pmol of each probe were used. An in vitro transcribed green fluorescent protein gene fragment was used as IC-RNA extraction control (2.5 pmol of IC-RNA–specific primers and 1.5 pmol of probe), as described above. 12 Cycling times were as follows: 1 cycle at 50°C for 30 min (reverse transcription), 95°C for 15 min, and 42 cycles at 95°C for 30 sec, 55°C for 30 sec, and 72°C for 30 sec. Pure water and a no-template control were used as negative controls in every run. Furthermore, WNV samples were comparatively analyzed using 3 previously published qRT-PCR assays (assays 3–5). Primer and probes of assay 3 16 targeted the WNV genome position 1160–1229, assay 4 17 the genome position 10–153, and assay 5 13 the genome position 10597–10672.
For the quantification of WNV copy numbers, a synthetic external calibrator was designed (Fig. 1A), which comprised the target sequences of assay 1 and used the same primers and probe. However, the binding site of the second probe (position 66–88) was mutated in this external calibrator by mirror inversions of 6 guanine/cytosine sequences to create a new specific target site, which can be detected only by a synthetic control probe (Fig. 1A; Table 1). An additional viral control probe (position 66–92) detected the corresponding original viral sequence. Using primer INEID f1 and INEID r1 in combination with the synthetic control probe allows the unambiguous detection of the synthetic control RNA, whereas the use of viral control probe together with assay 1 primers (INEID_f1 and INEID_r1) confirms viral RNA. This construct was amplified using vector PCR 2.1 g ; the vector was linearized with HindIII and in-vitro transcribed using a commercial in vitro transcription system. i The obtained transcripts were purified e (without carrier RNA), and the amounts of RNA were estimated. f
In order to determine the minimal copy number, an external calibrator was developed based on the WNV assay 1 target sequence and an authentic target site composition for the probe (Fig. 1A). Serial dilutions of this calibrator yielded copy numbers ranging from 1 to 2.5 × 10 7 copies/μl and were used to establish a calibration curve depicting mean threshold cycle (Ct) values plotted against the RNA copy numbers (Fig. 1B). The calibrator sequences were amplified in parallel using assay 1 primers and probes as well as assay 1 primers and synthetic control probe. The curve showed a linear progression for the WNV probe assay and a PCR efficiency of 1.0 and displayed (for synthetic control probe–derived assay) a PCR efficiency of 0.97. Both standard curves exhibited a correlation coefficient of >0.99. Based on this calibration curve, it can be concluded that the 2 new qRT-PCR assays are capable of detecting 2–4 RNA copies of WNV lineage 1 and 2 strains. The analytical sensitivity, as determined by the synthetic calibrator, is based on extractions from pure solutions. No inhibition was observed when this calibrator (100 RNA copies) was extracted from horse plasma (data not shown). In general, the impact of different matrix backgrounds (such as plasma or cell culture medium) was revealed by the internal control (present in all reactions and set to give a Ct value of 25–27). 12
The 2 novel qRT-PCR assays for WNV were compared to 3 previously published assays (assays 3–5) with regard to their sensitivity and amplification efficiency. To compare the analytical sensitivity of all assays, Ct values were normalized by comparing them with a positive RNA control (WNV strain NY99), which was added to each run. The experimental limit of detection (LOD) was set at the serial dilution corresponding to 3 copies of external calibrator, based on the finding that 3 copies per PCR reaction were detected to 100% (Fig. 1C). This is in accordance to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments Guidelines. 6
The analytical sensitivity comparison was carried out by determining duplicate Ct values of 10-fold dilutions (10–1–10–7) of WNV strain NY99-derived and Uganda-derived RNA (Table 2). The LOD for WNV strain NY99 was between 10–7 and 10–6 dilution, which corresponded to 1.2–12.2 copies per reaction. All assays detected the 10–6 dilution of NY99 RNA with similar analytical sensitivity: Assay 1 displayed a mean Ct of 34.3 ± 0, assay 2 a mean Ct of 34.5 ± 0, assay 4 a mean Ct of 34.5 ± 0.8, assay 5 a mean Ct of 34.8 ± 0.2, and assay 3 a mean Ct of 34.9 ± 0.2. In addition, assays 1, 3, and 5 were able to detect 10–7 RNA dilution (mean Ct of 36.1 ± 0.3 for assay 1, a mean Ct of 38.1 ± 0.6 for assay 5, and a mean Ct of 39.3 ± 1.0 for assay 3). The LOD for WNV strain Uganda was between 10–7 and 10–6 RNA dilution, which corresponded to 0.7–6.7 copies per reaction. The 10–6 Uganda RNA dilution was detected by assays 1 and 2 and harbored mean Ct values of 35.0 ± 0.1 and 37.3 ± 0.6, respectively. Assay 5 displayed a mean Ct of 38.6 ± 2.3, while assay 4 exhibited only a single Ct of 39.1. In addition, assay 1 detected 10–7 Uganda RNA dilution, with a mean Ct of 36.4 ± 0.4. Assay 3 is designed only for WNV lineage 1 strains. The results demonstrate that the new assays 1 and 2 are suitable for sensitive detection of both WNV lineages.
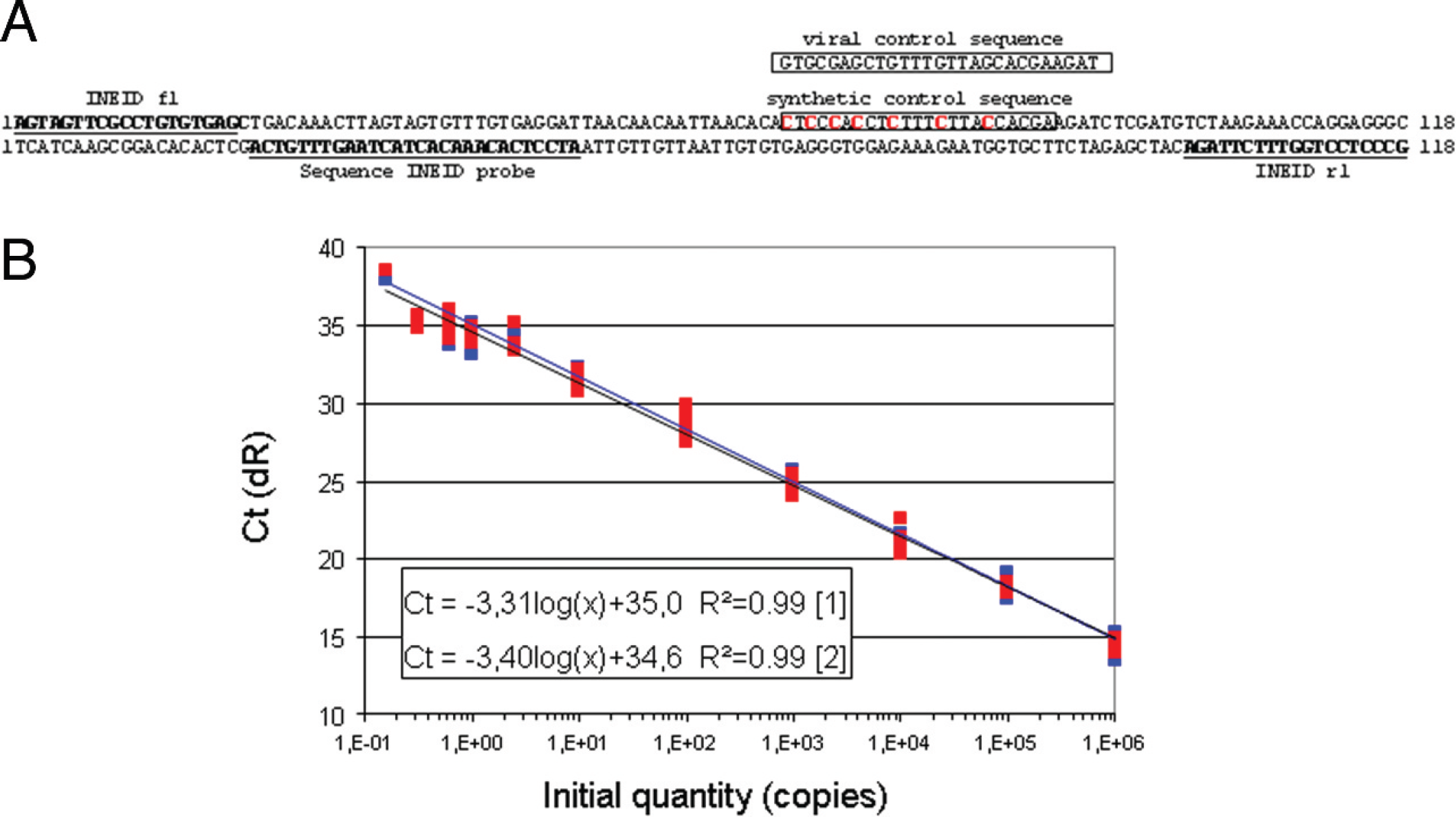
Synthetic calibrator.
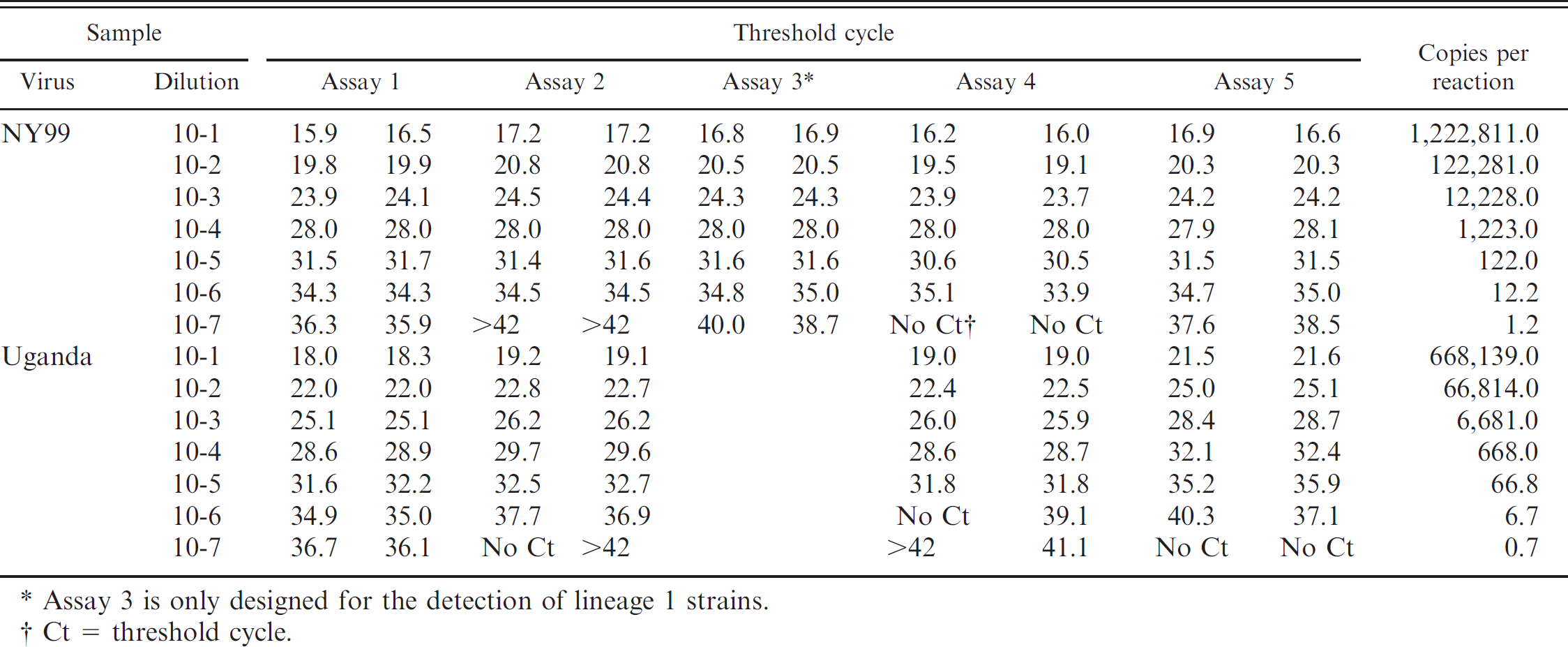
Assay 3 is only designed for the detection of lineage 1 strains.
Ct = threshold cycle.
Sensitivity of the real-time quantitative reverse transcription polymerase chain reaction assays for West Nile virus (WNV) strains NY99, Uganda 956, Kunjin, and Sarafend. *
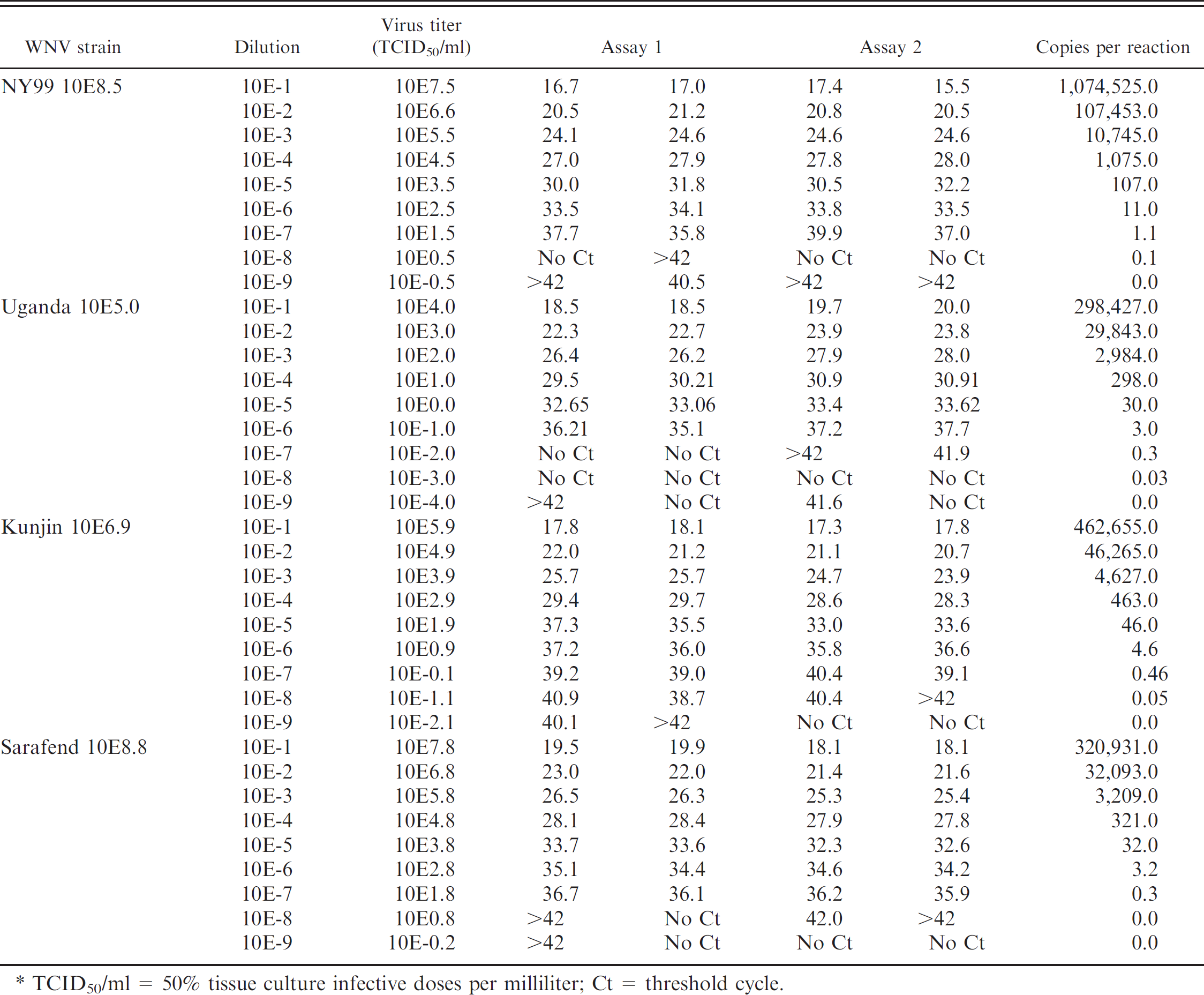
TCID50/ml = 50% tissue culture infective doses per milliliter; Ct = threshold cycle.
In addition, both of the new qRT-PCR assays were evaluated by testing the 4 WNV strains for which the infectivity titers in the tissue culture supernatants were determined: WNV strain NY99 contained 108.5 TCID50/ml, Uganda B956 contained 105.0 TCID50/ml, Kunjin contained 106.9 TCID50/ml, and Sarafend contained 108.8 TCID50/ml. The analytical sensitivity of each assay was evaluated by comparing duplicate Ct values of RNA extracts of 10-fold supernatant dilutions (Table 3). Both assays detected WNV strains of lineage 1 (NY99, Kunjin) and lineage 2 (Uganda B956, Sarafend) with an extremely high and comparable sensitivity. The LOD for WNV strain NY99 was in the range of 101.5–102.5 TCID50/ml, which corresponded to 1.1–11 copies per reaction. Assay 1 exhibited a similar sensitivity compared to assay 2, yielding a mean Ct of 36.75 ± 1.34 versus 38.45 ± 2.05 for a solution containing 101.5 TCID50/ml. The LOD for WNV strain Uganda was at a virus titer of 10–1 TCID50/ml, which corresponded to 3 copies per reaction. Again, both assays 1 displayed a comparable sensitivity (mean Ct of 35.66 ± 0.78 vs. 37.45 ± 0.35). For WNV strains Kunjin and Sarafend, the LODs were at a titer of 100.9 TCID50/ml (4.6 copies per reaction) and 102.8 TCID50/ml (3.2 copies per reaction). Even for these strains, assays 1 and 2 were compatible in terms of sensitivity for the detection of WNV strains Sarafend (mean Ct of 34.74 ± 0.52 vs. 34.40 ± 0.28) and Kunjin (mean Ct of 36.63 ± 0.86 vs. 36.2 ± 0.56).
Specificity of real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) assays for Murray Valley encephalitis virus (MVEV), Japanese encephalitis virus (JEV), Yellow fever virus (YFV), and Tick-borne encephalitis virus (TBEV). *
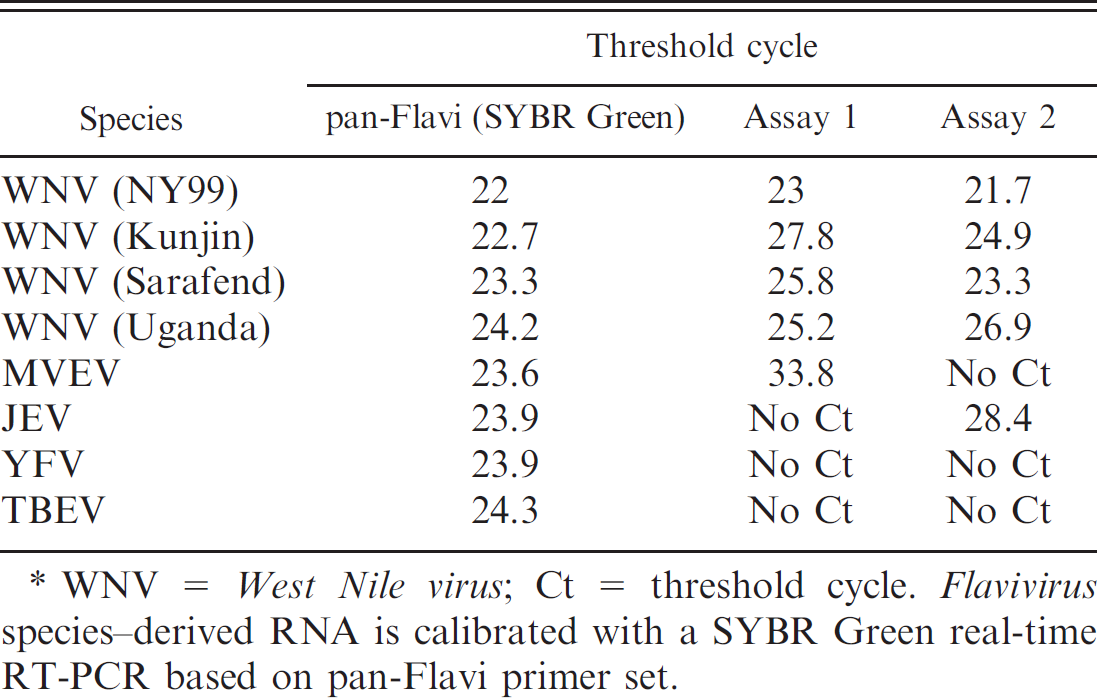
WNV = West Nile virus; Ct = threshold cycle. Flavivirus species–derived RNA is calibrated with a SYBR Green real-time RT-PCR based on pan-Flavi primer set.
The 2 new qRT-PCR assays were eventually tested with other species of genus Flavivirus, such as YFV, JEV, TBEV, and MVEV, in order to verify the specificity of the detection. For the adjustment of comparable RNA amounts for the different species, a SYBR Green real-time RT-PCR based on the pan-Flavi primer set (as previously published 7 ) was used, which targeted the conserved NS5 region of this genus. As shown in Table 4, assay 1 detected MVEV (Ct = 33.8), and assay 2 detected JEV (Ct = 28.4). All other analyzed species were not detected. Assay 2 may therefore be used under certain conditions for additional detection of JEV. Thus, the combined use of assays 1 and 2 helps one to avoid false-positive results for MVEV or JEV and enables the specific detection of WNV.
In summary, 2 new qRT-PCR assays were established for the detection of WNV genome, with excellent analytical sensitivity and good specificity. The high sensitivity of both assays for WNV lineage 2 allows the efficient detection of these emerging infections in Europe (i.e., Hungary since 2004 8 and Austria since 2008 [Department for Environment, Food Rural Affairs: 2008, West Nile virus: Austria. Reference: VITT 1200/WNV-Austria. Available at http://www.defra.gov.uk/foodfarm/farmanimal/diseases/monitoring/documents/wnv-austria.pdf. Accessed on April 15, 2010]). The 2 assays can be used in parallel or sequentially to mutually reconfirm the obtained results, since the 2 primer sets anneal at different regions of the WNV genome (highly conserved 5′UTR segment [assay 1] and NS2A region [assay 2]). Moreover, a new kind of external calibrator was designed, which relies solely on mirror-inversion mutations that can be discerned from the original viral sequences by a calibrator-specific RNA probe.
Acknowledgements
This work was funded by the Federal Ministry of Education and Research in the “Research on Zoonotic Infectious Diseases” program.
Footnotes
a.
Kindly provided by M. Niedrig, Robert-Koch-Institute, Berlin, Germany.
b.
Kindly provided by A. Muellbacher, John Curtin School of Medical Research, Canberra, Australia.
c.
Health Protection Agency, Salisbury, United Kingdom.
d.
Kindly provided by F. T. Hufert and M. Weidmann, Institute for Virology, Göttingen, Germany.
e.
QIAamp®, QuantiTect®, RNeasy® MinElute®; Qiagen GmbH, Hilden, Germany.
f.
Quant-IT™, Invitrogen Corp., Carlsbad, CA.
g.
Eurofins MWG Operon, Ebersberg, Germany.
h.
Mx3000P® QPCR system, Stratagene Inc., La Jolla, CA.
i.
Riboprobe®, Promega Corp., Madison, WI.