
Abstract
Toxoplasma gondii is a ubiquitous protozoan parasite that infects humans and many different animals, including felids. Many molecular and serologic tests have been developed for detection of T. gondii in a wide range of hosts. Loop-mediated isothermal amplification (LAMP) is a field-friendly technique that lacks the practical drawbacks of other molecular and serologic tests, and LAMP assays have been successfully developed for detection of T. gondii in fresh tissue samples. In the current study, both a previously published and a de-novo designed LAMP assay were compared to a quantitative real-time (q)PCR assay, for the detection of T. gondii in archived formalin-fixed, paraffin-embedded (FFPE) tissue samples from captive wildlife. The LAMP assays produced conflicting results, generating both false positives and false negatives. Furthermore, the LAMP assays were unable to positively identify samples with low levels of parasites as determined by qPCR and histopathology. Therefore, these LAMP assays may not be the most suitable assays for detection of T. gondii in archived FFPE and frozen tissue samples.
Toxoplasma gondii is a protozoan parasite that infects humans and a wide variety of animals (particularly felids), 7 and it is estimated that nearly one-third of the human population has been exposed to this parasite.3,5,11 Toxoplasma has been identified in more than 350 host species, mammals and birds, with the vast majority of them living in the wild. 25 Contamination of the environment is linked to the shedding of oocysts by stray, domestic, or wild felid species.5,25 However, there is a distinct lack of information regarding the prevalence of T. gondii in both captive and wild felids in South Africa.
Numerous molecular tests, including PCR and quantitative real-time (q)PCR, have been successfully developed and validated for detection of Toxoplasma in a wide variety of species.3,4,9 However, these tests require expensive laboratory equipment, reagents, highly trained professionals, and are generally time consuming in order to achieve accurate results.6,24 Furthermore, DNA isolated from fresh or frozen tissues are generally most suitable for these molecular assays. However, formalin-fixed, paraffin-embedded (FFPE) tissues are routinely used by pathologists for histologic diagnosis of disease and to detect pathogens in human beings and wildlife. Hence, FFPE tissues are often the only sample type available for molecular analysis, particularly for retrospective studies. Although archival samples, such as FFPE tissues, are invaluable sources for analysis, the fixation process causes cross-linking of DNA, RNA, and proteins within cells.16–18 Nucleic acids can successfully be purified from these tissues but these nucleic acids are generally fragmented, and amplification of fragments >500 base pairs (bp) is seldom possible via PCR. 12
Loop-mediated isothermal amplification (LAMP) is an alternative nucleic amplification technique that does not require expensive equipment, laboratories, reagents, or highly trained personnel.8,23 The LAMP technique facilitates rapid, qualitative diagnosis and thus may be more useful as a detection tool than PCR or qPCR, particularly in resource-poor areas or in the field. 8 The LAMP assay uses a single DNA polymerase with strong strand displacement activity and a set of 4–6 specially designed primers facilitating rapid isothermal (usually at 63°C) amplification of a DNA or RNA nucleic acid target. Positive results can be identified visually by turbidity 19 or addition of fluorescent DNA-binding dyes. 23 The LAMP technique has been successfully used in several studies for molecular detection of numerous bacterial, fungal, protozoal, and viral infections,6,20,26 and there are several commercial kits available for detection of pathogens such as Cryptosporidium, Escherichia coli O157, Giardia, Legionella, and Salmonella among others. A range of LAMP assays have also been successfully developed for detection of pathogens in FFPE samples, for example, for Penicillium marneffei 28 and Mycobacterium tuberculosis. 30
LAMP assays have also been published for detection of Toxoplasma in fresh tissues.3,13,34 However, to date, no LAMP assay has been described for detection of T. gondii in archived FFPE and/or frozen tissue samples of captive felids, to our knowledge. As LAMP assays usually produce target amplicons 100–300 bp in length, which is similar in size to qPCR and generally smaller than PCR amplicons, it was thought that a smaller target amplicon length may improve the capability of LAMP for detection of T. gondii in archived tissues. Furthermore, the 529-bp repeat element sequence is found ~300 times within the T. gondii genome and has successfully been used in several PCR and qPCR assays for detection of toxoplasmosis.4,15,29 Therefore, it was hypothesized that the small amplicon length and multifold copies of the 529-bp repeat element sequence would facilitate a more sensitive LAMP assay than PCR from archived tissue samples.
The aim of this study was to compare the performance of 2 different primer sets for LAMP, which target the 529-bp repeat element sequence, for detection of T. gondii in FFPE and frozen tissues obtained from captive felids. One assay used primers from a previously published LAMP assay used to identify T. gondii infections from necrotic lesions and blood in pigs15,34 and in blood from children with leukemia, 9 whereas the second assay used novel primers designed in this study.
A total of 22 tissue samples (13 FFPE and 9 frozen) from 13 African black-footed cats (Felis nigripes), 5 cheetahs (Acinonyx jubatus), and 4 African hunting dogs (Lycaon pictus) were obtained from the NZG Biobank, National Zoological Gardens of South Africa, South Africa. Histopathologic data was used to initially select samples for this study. Six F. nigripes samples and 1 A. jubatus sample showed typical lesions associated with characteristic T. gondii tissue cysts. Five samples from F. nigripes and 4 from A. jubatus with typical lesions in which T. gondii cysts could not be detected were used to test if the LAMP assay could be used to confirm the suspected pathology diagnosis. Two samples from F. nigripes and 4 from L. pictus that were not infected with Toxoplasma were used as negative controls. The FFPE samples were comprised of blocks, with each block containing a mixture of organs, including brain, heart, kidney, liver, and lung. Frozen samples were comprised of liver tissue, with the exception of sample 12Z035, which was a spleen sample.
A previously published qPCR assay 4 targeting the B1 gene of T. gondii was used to compare with the histologic data. This B1 qPCR assay has been shown to be specific for Toxoplasma and does not cross-react with closely related species such as Neospora. 4
Purified genomic DNA from T. gondii (strain I LDM) was obtained from the National Institute of Communicable Diseases (South Africa) and used as a positive control for this study. Genomic DNA was isolated from FFPE sections and frozen tissue samples using a commercially available kit a according to the manufacturer’s instructions. The DNA concentration was measured using a spectrophotometer, b and all samples were diluted to a standard DNA concentration of 10 ng/µL.
Two sets of LAMP primers targeting the 529-bp repeat element of T. gondii (GenBank accession AF146527) were used for the LAMP assays (Table 1). Primer set 1 (referred to as LAMP_Z) consisted of 6 previously published oligonucleotide primers.9,15,34 Primer set 2 (referred to as LAMP_ES) was designed from the complete 529-bp repeat element (accession AF146527) using LAMP primer design software. c
Loop-mediated isothermal amplification primers used in the current study.*

FIP = forward internal primer; BIP = backward internal primer.
The LAMP reaction is extremely sensitive, thus small amounts of contamination can result in false positives. Therefore, stringent quality assurance measures and controls were implemented for all LAMP assays performed in this study. Each reaction tube was prepared (using sterile filter tips) and sealed separately in a class II biological safety cabinet prior to LAMP. Furthermore, LAMP reactions were performed in a dedicated PCR laboratory and gel electrophoresis in a separate laboratory to further minimize risk of contamination. Negative controls (water blank) consistently failed to amplify even in repeated runs.
Genomic DNA from T. gondii (strain I LDM) was used for validation of both LAMP assays, which were performed as described previously. 34 Briefly, the LAMP reaction (25 µL) contained 50 ng of template DNA, 40 pmol each of the inner primers (FIB and BIP), 5 pmol of outer primers F3 and B3, 20 pmol of loop primers F2 and B2, 8 U of Bsm DNA polymerase, b 1.4 mM deoxyribonucleotide triphosphates, and 2× reaction buffer (1.6 M betaine, 40 mM Tris–HCl [pH 8.8], 20 mM KCl, 20 mM (NH4)2SO4, 16 mM MgSO4, and 0.2% Tween 20 d ). Reactions were performed at 63°C for 1 h in a thermocycler. e To determine the sensitivity of both assays, LAMP reactions were also performed on a 10-fold dilution series (100 ng to 0.1 pg) of purified T. gondii strain I LDM DNA. Thereafter, 10 µL of the reaction products were electrophoresed on a 1.5% agarose gel and stained. f Additionally, 1 µL of stain f was added to the remaining 15-µL reaction mixture, and then left for a few minutes at room temperature. Fluorescence in the tubes was visualized using an ultraviolet transilluminator to identify positive results (Supplementary Fig. 1, available at http://vdi.sagepub.com/content/by/supplemental-data; Fig. 1). The LAMP assays using the LAMP_Z and LAMP_ES primer sets successfully amplified DNA isolated from the positive control, T. gondii strain I LDM (Fig. 1A). Furthermore, the LAMP_ES primer set developed in this study had comparable performance and sensitivity to the previously published LAMP_Z primer set 34 with both assays having a detection limit of 1 pg purified DNA (Fig. 1A).
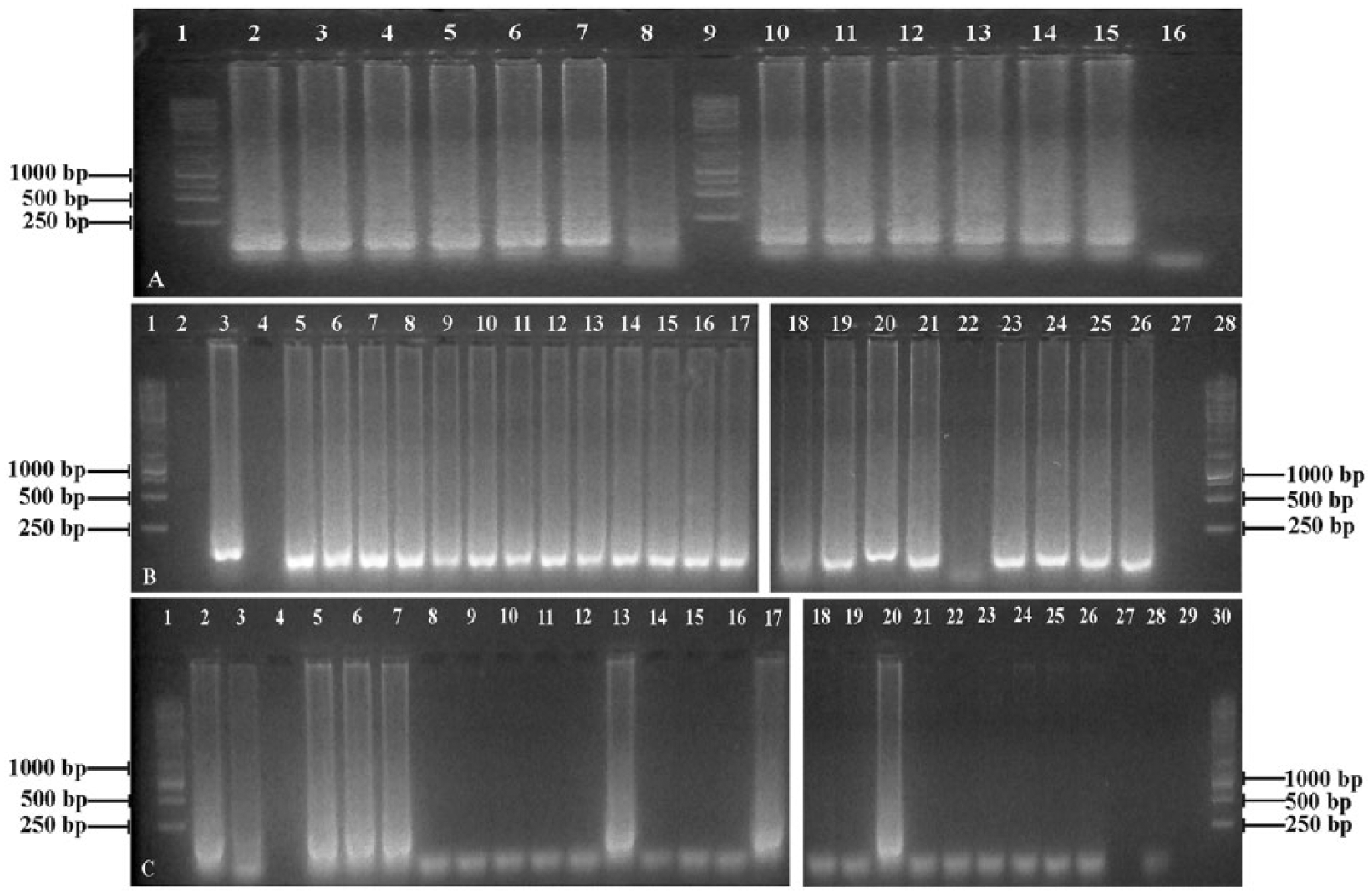
Results of loop-mediated isothermal amplification (LAMP) assays for detection of Toxoplasma gondii.
The LAMP_Z and LAMP_ES primers sets were used to detect the presence of T. gondii in captive felids from archived FFPE and frozen tissue samples; LAMP assays were performed and visualized as described above. Positive results appeared as smears after electrophoresis (Fig. 1). Generally, positive LAMP results can be identified by a ladder-like banding pattern after agarose gel electrophoresis but may also appear as smears.1,31 Twenty-one samples tested positive (95%) when tested with the LAMP_ES primer set, whereas only 27% of the samples tested positive when tested with the LAMP_Z primers (Table 2). The results obtained with the LAMP_ES and LAMP_Z primer sets were non-concordant and inconsistent compared to pathologic findings and qPCR (Fig. 1B, 1C; Table 2), suggesting that the LAMP_ES primer set resulted in numerous false positives, whereas the LAMP_Z primer set produced several false-negative results (Fig. 1B, 1C; Table 2). Several other publications have anecdotally reported false-positive results with the LAMP technique, including assays for detection of Plasmodium falciparum malaria, 27 African trypanosomes, 14 and Salmonella.32,33 The LAMP assays were repeated on those samples that generated conflicting results with both sets of primers (Fig. 2; Table 2) and resulted in additional inconsistencies: samples that were negative with the LAMP_Z primers in the first run were positive in the second run (Fig. 2; Table 2).
Summary of results for the loop-mediated isothermal amplification (LAMP) assays with primer sets LAMP_ES and LAMP_Z, including digests with TaqI and TfiI.*
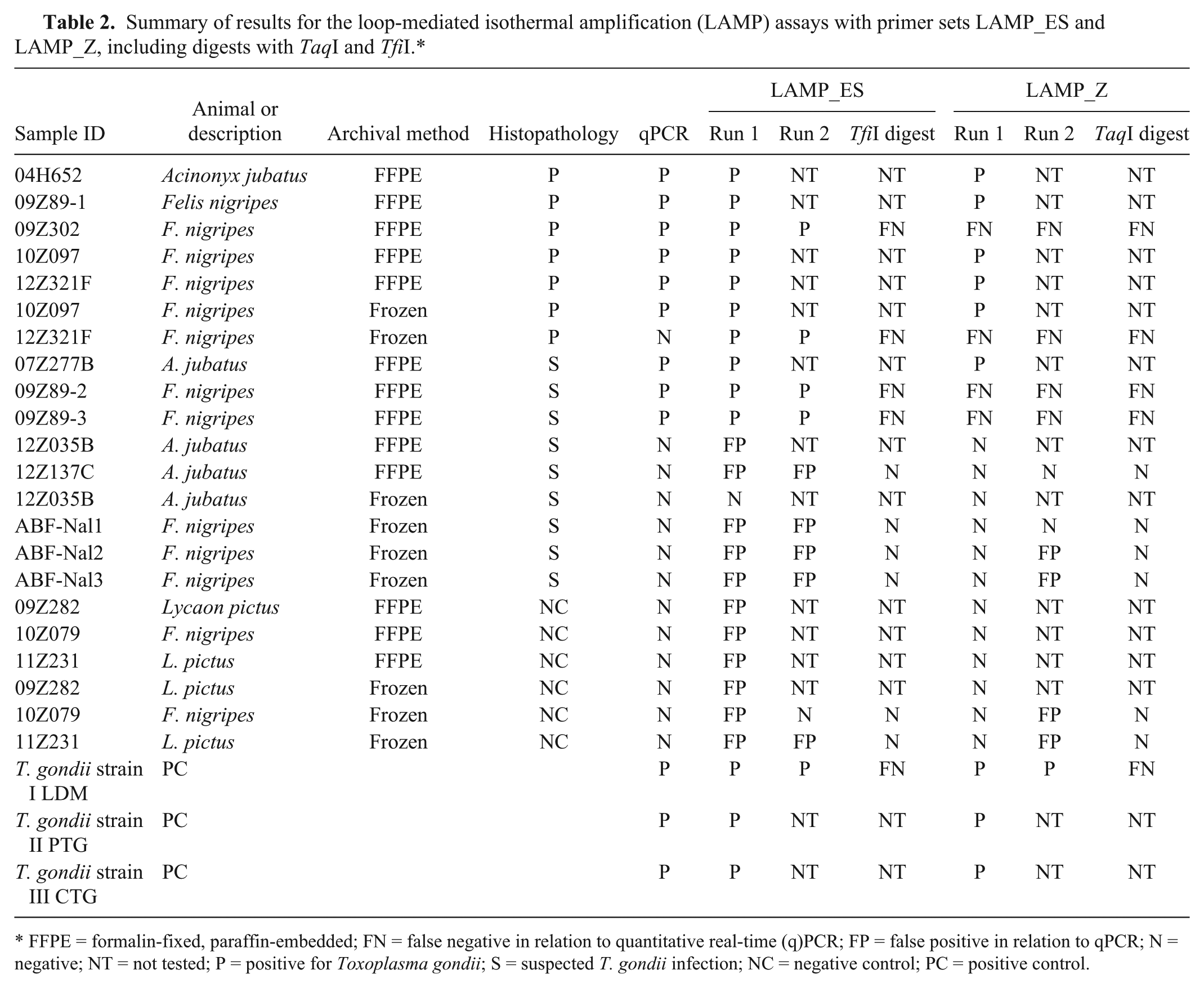
FFPE = formalin-fixed, paraffin-embedded; FN = false negative in relation to quantitative real-time (q)PCR; FP = false positive in relation to qPCR; N = negative; NT = not tested; P = positive for Toxoplasma gondii; S = suspected T. gondii infection; NC = negative control; PC = positive control.
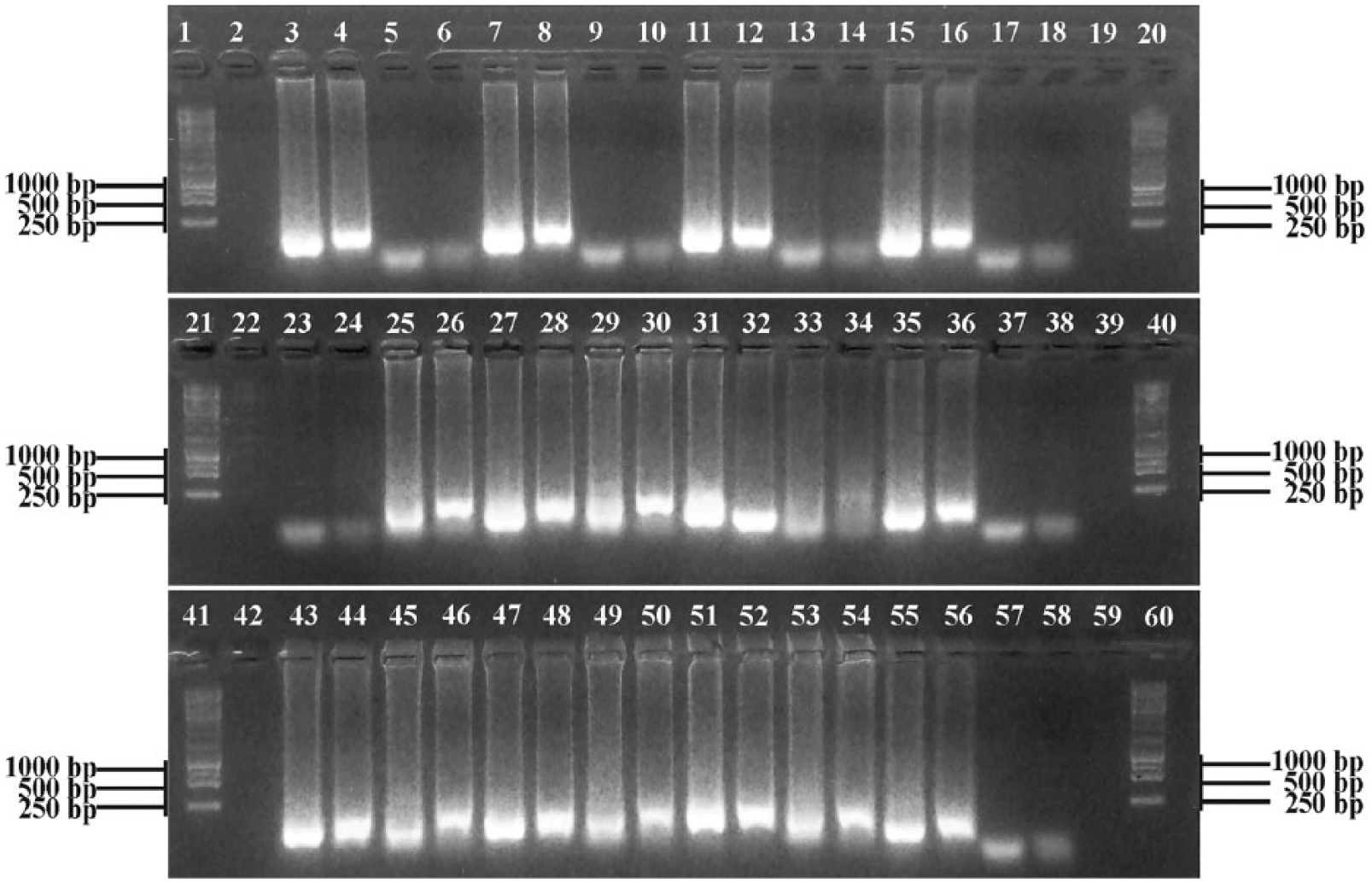
Repeat of loop-mediated isothermal amplification (LAMP) assay to determine false-positive and false-negative results by restriction analysis of LAMP products. LAMP_ES assay results: lanes 3, 7, 11, 15, 23, 27, 31, 35, 43, 47, 51, and 55. Restriction digestion of LAMP_ES amplicons with TfiI: lanes 4, 8, 12, 16, 24, 28, 32, 36, 44, 48, 52, and 56. LAMP_Z assay results: lanes 5, 9, 13, 17, 25, 29, 33, 37, 45, 49, 53, and 57. Restriction digestion of LAMP_Z amplicons with TaqI: lanes 6, 10, 14, 18, 26, 30, 34, 38, 46, 50, 54, and 58. Lanes 1, 20, 21, 40, 41, 60: 1-kb DNA ladder f ; lanes 2, 19, 22, 39, 42, 59: blank; lanes 3–6: 09Z89-2; lanes 7–10: 09Z89-3; lanes 11–14: 09Z302; lanes 15–18: 12Z137C; lanes 23–26: 10Z079; lanes 27–30: 11Z231; lanes 31–34: 12Z321F; lanes 35–38: ABF-Nal1; lanes 43–46: ABF-Nal2; lanes 47–50: ABF-Nal3; lanes 51–54: Toxoplasma gondii strain I LDM; lanes 55–58: negative control.
A key feature of LAMP is an absence of the need for expensive equipment and highly trained laboratory personnel, particularly advantageous in rural and field environments. Direct sequencing of LAMP products has been used to confirm positive results 10 but this is not an ideal tool for confirmation of LAMP results because LAMP amplicons are small and the products form long stem-loop repetitive structures. Furthermore, sequencing of LAMP products is costly and requires expensive laboratory equipment and highly trained staff. Several studies have reported confirmation of LAMP results by restriction digests based on naturally occurring restriction sites occurring within the LAMP target sequence.2,21,22,26 Restriction digestion is a simple, quick, and cheap alternative to sequencing of LAMP products that can be used to confirm positive results and may be used for genotyping. The same equipment used for LAMP can be used for restriction analysis, facilitating confirmation of results even in rural and field environments.
In silico analysis of the 529-bp repetitive element sequence indicated the presence of a TaqI restriction site within the target sequence amplified by the LAMP_Z primers and the presence of a TfiI restriction site within the target sequence amplified by the LAMP_ES primers. Therefore, the LAMP assays were repeated, and 10 µL of the products from the LAMP_Z and LAMP_ES primer sets were digested (separately) with TaqIb and TfiI,b respectively, according to the manufacturer’s instructions, for 1 h. The undigested and digested LAMP products were then electrophoresed on a 1.5% agarose gel (Fig. 2) and stained. Restriction digests failed to facilitate identification of true-positive results when compared to histologic and qPCR data (Fig. 2; Table 2). These results indicate that, despite restriction digestion, both LAMP_Z and LAMP_ES primer sets were unsuitable for detection of T. gondii in wildlife from archived FFPE and frozen tissue samples by LAMP.
Although the LAMP technique has many advantages compared with techniques such as PCR and qPCR, the 2 primer sets used in our study may not be the most appropriate assay for detection of T. gondii in wildlife from archived FFPE and frozen tissue samples. This could be attributed to partial hybridization of 1 or more LAMP primers to fragmented genomic host DNA, which may have then been randomly amplified by the Bsm DNA polymerase used for LAMP. In silico hybridization analysis of the primer sequences used in this study showed partial homology to genomic DNA sequences of both F. nigripes and A. jubatus. The problem of false positives associated with the LAMP primers used in this study highlights the importance of confirmation and verification of results obtained by the LAMP technique.
Footnotes
Acknowledgements
We thank Prof. John Frean and Desire du Plessis at the Centre for Opportunistic, Tropical and Hospital Infections, National Institute for Communicable Diseases, South Africa, for providing purified Toxoplasma gondii DNA, which were used as controls in the study.
Authors’ contributions
E Suleman contributed to conception and design of the study; contributed to acquisition, analysis, and interpretation of data; and drafted the manuscript. MS Mtshali contributed to conception and design of the study; contributed to acquisition, analysis, and interpretation of data; critically revised manuscript; and gave final approval. E Lane contributed to acquisition, analysis, and interpretation of data; critically revised manuscript; and gave final approval. All authors agreed to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
ZR FFPE DNA MiniPrep isolation kit, ZR Genomic DNA–Tissue MiniPrep kit; Zymo Research, South Africa.
b.
NanoDrop ND-1000 spectrophotometer, Bsm DNA polymerase, FastDigest TaqI restriction endonuclease, FastDigest TfiI restriction endonuclease, FastDigest EcoRI restriction endonuclease; Thermo Fisher Scientific, South Africa.
c.
Eiken PrimerExplorer V4 software, Eiken Chemical Co. Ltd., Tokyo, Japan.
d.
Tween 20, Sigma-Aldrich Pty. Ltd., Johannesburg, South Africa.
e.
T-100 thermo cycler, Bio-Rad Laboratories Ltd., Johannesburg, South Africa.
f.
GR Green dye, O’Gene Ruler; Thermo Fisher Scientific, South Africa.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was financed by the National Research Foundation of South Africa and the National Zoological Gardens of South Africa, under the research grant allocated to E. Suleman.