
Abstract
A multiplex (m)PCR and a PCR followed by restriction fragment length polymorphism (RFLP) analysis of Avibacterium paragallinarum have been proposed as alternatives to conventional serotyping by the Page scheme. We evaluated both methods, and also sequenced the PCR-RFLP target fragment to reexamine the capacity of molecular serotyping. Eleven reference strains and 27 field isolates were used. Many reference strains and isolates were misidentified as Page serogroup B. The sequence analysis revealed 6 profiles based on the matching rates of the target sequence with the 3 reverse primers of the mPCR. The reference strains and field isolates in profiles 1 and 4 were correctly identified as serogroup A or C by the mPCR. The strains and/or isolates in profiles 2, 3, 5, and 6 could be misidentified as serogroup B or as nontypeable by the mPCR. The homology comparison of the sequences showed that the target sequence of the mPCR, called region 2, was not Page serogroup specific, although some Kume serovars, such as A-1 and C-2, were correctly serotyped. In addition, there was a 9 nucleotide deletion in the sequences of profiles 1, 3, and 5, but not of profiles 2, 4, and 6. Overall, we confirmed that the mPCR and PCR-RFLP molecular assays are not suitable for identifying the serogroups of A. paragallinarum isolates. With further study, analysis of region 2 sequences may have potential as a means of recognizing the Kume serovars of A. paragallinarum isolates.
Keywords
Infectious coryza, caused by Avibacterium paragallinarum, is an important respiratory disease of chickens that causes economic losses wherever chickens are raised. 1 There are 2 schemes for A. paragallinarum serotyping, the Page scheme (which recognizes serogroups A–C) and the Kume scheme (which recognizes serogroups A–C and then serovars A-1, A-2, A-3, A-4, B-1, C-1, C-2, C-3, and C-4), and both schemes use the hemagglutination inhibition (HI) test.1–3 The HI test is demanding as it requires high titer rabbit antisera, chicken erythrocytes fixed with glutaraldehyde, and treatment of antigens to ensure hemagglutination activity. For the Kume scheme, the assay is even more demanding, requiring complex absorption procedures to produce serovar-specific antisera. Thus, the capacity to serotype A. paragallinarum, particularly by the Kume scheme, is restricted to just a few laboratories around the world. 1
Although serotyping is demanding and difficult, knowledge of the Page serogroups present in a geographic region is critical in the use of vaccines, as inactivated vaccines only provide protection against the Page serogroup present in the vaccines. 1 There is also evidence that, within a Kume serogroup, not all serovars will provide cross-protection. 9 Hence, the limited availability to serotype A. paragallinarum isolates is a major restriction to the effective prevention and control of infectious coryza outbreaks by vaccination.
Various PCR-based serotyping methods have been developed for several bacterial pathogens, including members of the family Pasteurellaceae, such as Actinobacillus pleuropneumoniae, 10 Haemophilus influenzae, 11 and Pasteurella multocida. 5 For A. paragallinarum serogrouping by the Page scheme, a multiplex (m)PCR and a PCR–restriction fragment length polymorphism (RFLP) method have been developed. 8 The assays are based on a hypervariable region of the HMTp210 gene, also referred to as region 2, encoding the protective hemagglutinin (HA) protein.8,12 Although the concept of these assays is attractive, a 2014 evaluation of mPCR with 12 reference strains and 69 field isolates from the Americas showed a relatively poor performance. 6 In the current study, we evaluated the mPCR and the PCR-RFLP assays using 11 reference strains and 27 isolates from various origins, the latter obtained since 1985 (Table 1). Because the current evaluation results also revealed some discrepancies between serotyping and mPCR results, in agreement with the previous report, 6 the target fragments were sequenced and analyzed.
Origin, Page serogroup, Kume serovar, multiplex (m)PCR, and PCR–restriction fragment length polymorphism (PCR-RFLP) results for the Avibacterium paragallinarum reference strains and isolates included in the current study.*
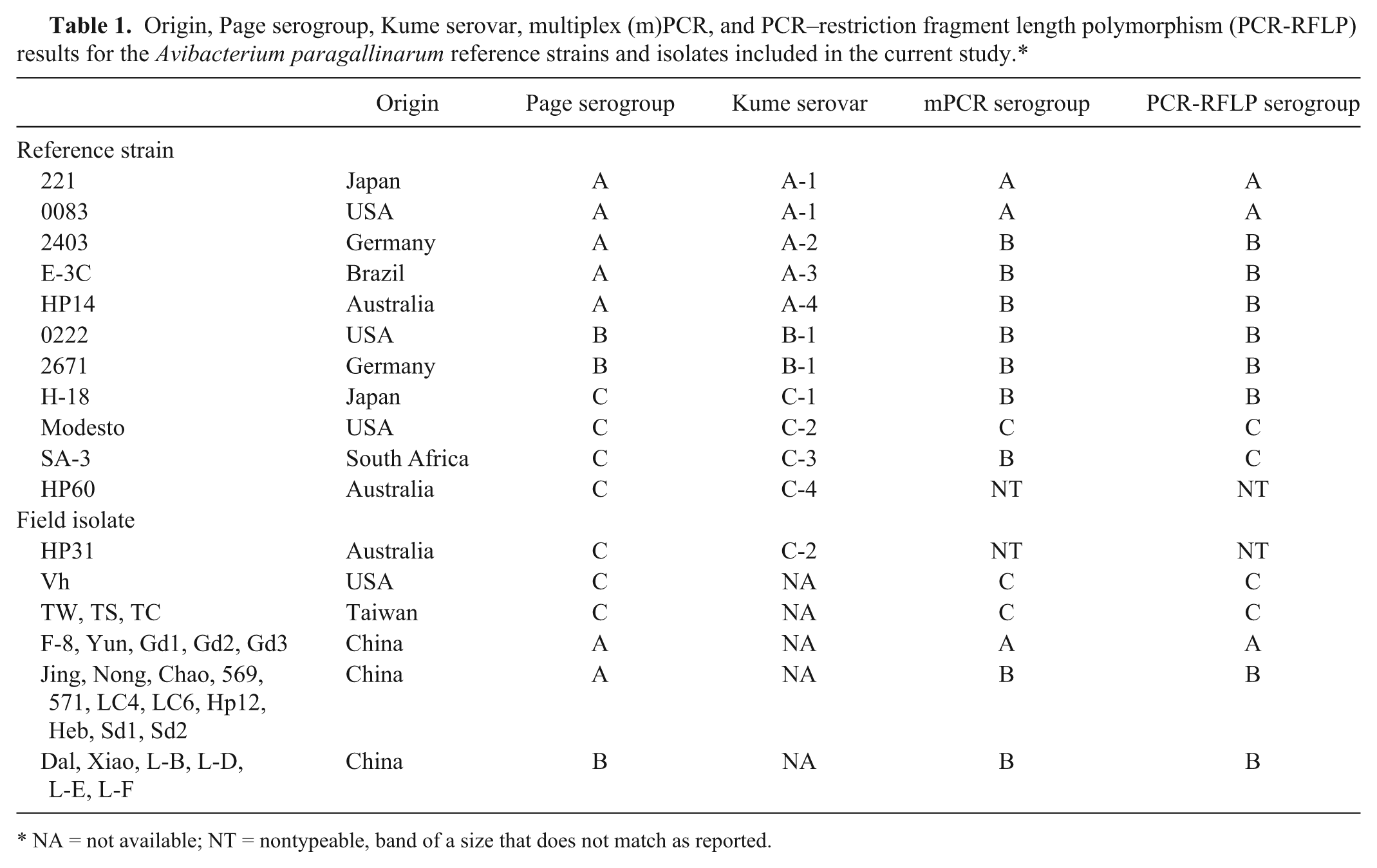
NA = not available; NT = nontypeable, band of a size that does not match as reported.
The identity of all strains and isolates was confirmed by a species-specific PCR. 4 The field isolates were allocated to a Page serogroup using the described methodology and antisera produced in China to the 3 Page serogroup reference strains. Those isolates that showed a disagreement between the HI test and the mPCR result were reexamined using reference antisera produced in Australia. a The mPCR and PCR-RFLP were conducted as described previously. 8 All PCR products for RFLP were sequenced b using ⊿5-1 forward and ⊿5-1 reverse 8 as sequencing primers, and the sequence data were analyzed using a commercial package. c
The mPCR results for the reference strains fully agreed with a recent evaluation (Table 1). 6 Of the 27 field isolates, 5 of 16 serogroup A, all 6 serogroup B, and 4 of 5 serogroup C isolates were correctly assigned into serogroup A, B, or C. The remaining 11 serogroup A isolates were recognized as serogroup B, and 1 Page serogroup C/Kume serovar C-2 isolate (HP31), which amplified the same size band as reference strain HP60 (about 0.4 kb), was nontypeable by the mPCR (Table 1).
To compare the performance of the mPCR in the current study with the prior evaluation, 6 we directly compared the studies (Table 2). We accepted the conventional serotyping as the correct answer and have shown, within each serogroup, the total correct and erroneous results. We did not include any results in which the mPCR failed to give the expected serogroup A, B, or C product (i.e., NT), of which there were 2 in our study and 3 in the prior study. 6 This direct comparison shows that both studies found that the mPCR gave correct results with some strains/isolates across all 3 Page serogroups. Furthermore, both studies found that most problems occurred when the mPCR gave a serogroup B result, with 16 of the 24 serogroup B results in the current study being incorrect and 18 of the 29 in the earlier study. 6 However, when examined in greater detail, the earlier study found that most false serogroup B mPCR results were associated with serogroup C (15/18), 6 whereas in our study the false mPCR serogroup B results were mainly associated with serogroup A (14/16). The different geographic sources of the 2 studies, mainly China for our study as compared with Central and South America for the earlier study, 6 could be a contributing factor for the difference in results. It is unclear what further problems with erroneous results might occur if strains from other regions were to be examined by the mPCR.
Comparison of multiplex PCR results given in the current study and the Morales-Erasto et al. study. 6

PCR-RFLP results showed that the method only surpassed the mPCR by correctly identifying reference strains SA-3 (C-3) as serogroup C. Both Australian strains, HP60 (Kume serovar C-4 reference strain) and HP31 (Kume serovar C-2 field isolate), amplified a 1.75-kb band and were digested by BglII to yield 1,469- and 289-bp bands. The serogrouping result for other strains and/or isolates was the same as the mPCR (Table1). The strains that were assigned to serogroup B by the RFLP showed no BglII restriction site in their RFLP-PCR sequences.
Given that primers are the key factor for the specificity of a PCR assay, the PCR product for RFLP 8 of all 38 strains and/or isolates was sequenced, and each sequence was compared with the 3 mPCR reverse primers. The region 2 sequences of serogroup A 221 (GenBank AR303123) and serogroup C 53-47 (AR698445)7,8 were included in the analysis. The sequence alignment revealed 6 profiles. These profiles were based on the matching rate with the A, B, and C reverse primers and were as follows: profile 1—100%, 60%, and 93.3%; profile 2—66.7%, 100%, and 100%; profile 3—66.7%, 100%, and 90%; profile 4—66.7%, 90%, and 100%; profile 5—66.7%, 100%, and 93.3%; profile 6—95.2%, 100%, and 100% (Table 3).
The matching rates of region 2 sequences with the reverse primers for Page serogroups in the multiplex (m)PCR.*
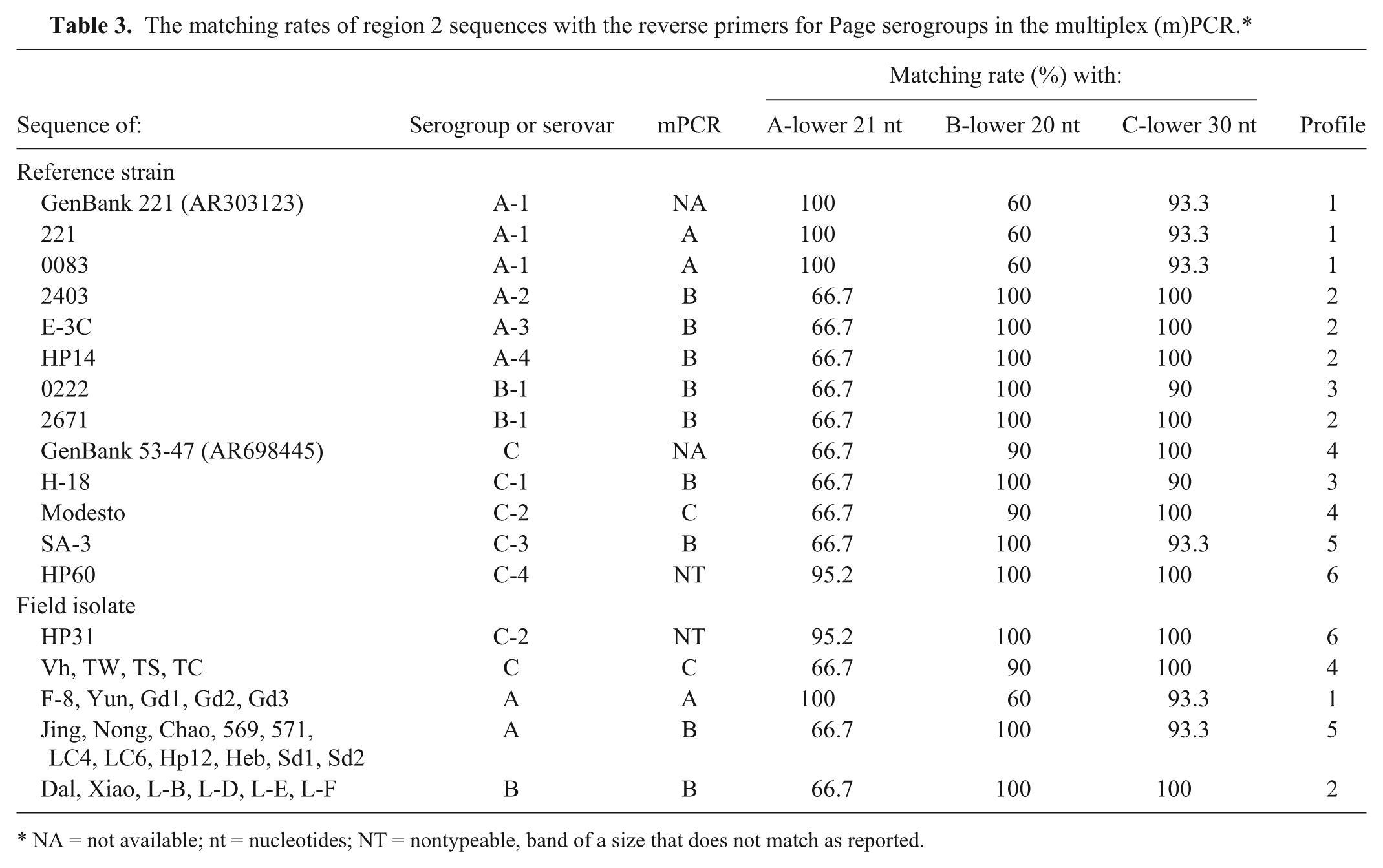
NA = not available; nt = nucleotides; NT = nontypeable, band of a size that does not match as reported.
The target sequences in profiles 1 and 4, generated with serovar A-1 and C-2 reference strains as well as 5 serogroup A and 4 serogroup C field isolates, were 100% matched only with the corresponding A or C primer and so these strains were correctly serogrouped by mPCR. The sequences in profiles 3 and 5 (representing serovar B-1 (0222), C-1, and C-3 reference strains, and 11 serogroup A isolates) were well matched with the designed B reverse primer, and were identified as serogroup B by mPCR. The sequences in profile 2 (representing serovar A-2, A-3, A-4, and B-1 (2671) reference strains, and 6 serogroup B field isolates) were 100% matched with both the B and C reverse primers, and were also identified as serogroup B by mPCR. The sequences of profile 6, the serovar C-4 reference strain HP60, and the serovar C-2 isolate HP31 (both are Australian sourced), which were 100% matched with both the B and C reverse primers, and were 95.2% matched with the A reverse primer (with only 1 mismatch near the 5′-end of the primer), produced a 0.4-kb band. The 0.4-kb band was sequenced and found to be the amplicon of the mPCR ABC forward primer and A reverse primer. This situation might imply that a shorter fragment was favored to be amplified in the mPCR at given conditions. Given that the serogroup B reverse primer showed a 100% match with 7 of 9 serovar reference strains, it is not surprising that 5 serovar reference strains and 11 serogroup A isolates were misidentified as Page serogroup B (Table 3).
For accurate nucleotide alignment and amino acid translation of the region 2 sequence, a second-round PCR product sequencing was conducted for all 38 strains and/or isolates using the mPCR ABC forward primer (which was 90 nucleotides [nt] ahead of ⊿5-1 forward primer) and ⊿5-1 reverse primer 8 to amplify a 1.7-kb fragment. Two sequencing reactions were then performed. The first sequencing was done using the ABC forward primer. The second sequencing was done using a newly designed primer p1540 (5′-AGTAATGTCGCCGATGG-3′, at position ~1,540 nt from the ABC forward primer and 160 nt to the end of ⊿5-1 reverse primer). The 2 sequences were used to trim the previous sequences to obtain complete region 2 sequences starting from ⊿5-1 forward primer and ending at ⊿5-1 reverse primer. The 38 sequences were submitted to GenBank with accessions KU143734–KU143744 and KU167070–KU167096. The homology of these sequences was compared using a commercial software package, c and a phylogenetic tree was drawn with a commercial software package (Fig. 1). d The sequence similarity within serogroup A varied from 53.0% to 100%, but within serovar A-1 and 5 of 16 field serogroup A isolates was 99.8–100%. The sequence similarity within serogroup B or serovar B-1 was 99.2–100%. The sequence similarity within serogroup C was 96.5–100%, and within serovar C-2 Modesto and 5 of 6 field serogroup C isolates, including 53-47 (AR698445), was 99.9–100%. Australian isolate HP31, a serovar C-2 strain, held 96.5–96.7% sequence similarity with Modesto and 5 other serogroup C isolates, but shared 99.9% sequence similarity with serovar C-4 HP60. According to the sequence data, region 2 was highly variable (53.2–56.7% in similarity) between serovar A-1 and the other 8 Kume serovars. Among these 8 serovar reference strains, however, the sequence identity was over 90%. The detailed homology is presented as Supplemental Table 1 (available at http://vdi.sagepub.com/content/by/supplemental-data).
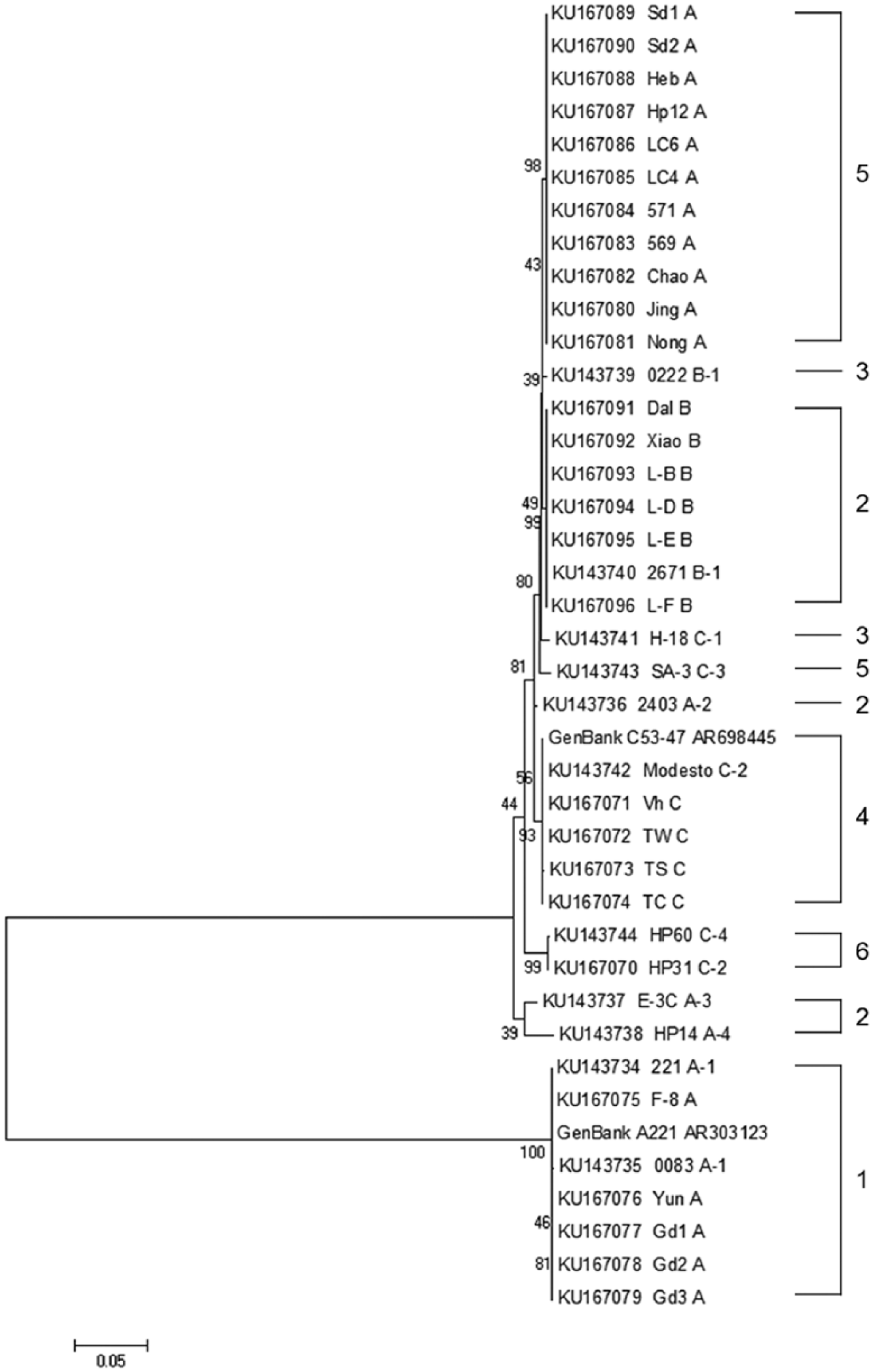
The phylogenetic relationship based on neighbor-joining analysis of region 2 sequences from 11 Avibacterium paragallinarum reference strains, 27 field isolates, and 2 GenBank control sequences. The numbers at nodes indicate bootstrap value obtained from 1,000 resamplings. The scale bar represents sequence variation.
Sequence analysis found that there was a repeat of 9 nt—GTATTGATG/GTATTGATG (at position 1427-1444 nt of strain 2671)—in the sequences of the Kume serovar A-2, A-3, A-4, B-1 (2671), C-2, and C-4 reference strains, and field isolates of Page serogroup B and C (i.e., profiles 2, 4, 6), but second GTATTGATG was missing in the same area of Kume serovar A-1, B-1 (0222), C-1, and C-3 reference strains, and 11 field isolates of Page serogroup A (i.e., profiles 1, 3, 5). The 9 nt status in 0222 and H-18 (profile 3) was slightly different, where the first “G” was replaced by “A.”
With the proposed mPCR, it is impossible to distinguish the strains and/or isolates with profiles 2, 3, and 5, which contains 24 of 38 strains and/or isolates used in the study. However, these same strains and/or isolates might be discriminated at serovar level by examining a few features within the region 2 sequences (i.e., a BglII restriction site, the profile type, sequence homology [>99.2% identity within a serovar], and 9 nt deletion status). The region 2 sequence of the 11 Page serogroup A field isolates with profile 5 were 100% identical and most closely related to Page serogroup B reference 0222 (profile 3) with 99.3% similarity and 9 nt deletion. The 11 isolates had been suspected as serogroup B, but repeated HI tests determined that they were truly serogroup A. The Kume serovar B-1 reference strain 2671 (profile 2), which was 99.3% identical with the 11 serogroup A isolates (profile 5), had no 9 nt deletion. In contrast, the 6 isolates of serogroup B (profile 2) had 99.9% similarity with reference strain 2671 and no 9 nt deletion, as well. These results suggest that the 6 Chinese field isolates of serogroup B (profile 2) could be called 2671-like serogroup B isolates. Both mPCR and the region 2 sequence analysis failed to allocate the 11 serogroup A field isolates into any pattern found within the existing Kume serovar reference strains. Whether or not these isolates belong to a new Kume serovar needs full conventional Kume serotyping and further study. Our results suggest that Page serogroup B may be made up of more than 1 Kume serovar. The Page serogroup B reference strain 0222 has been shown to be Kume serovar B-1. However, the Kume serovar B-1 reference strain 2671 differed from the Page serogroup B reference strain (0222) in profile type and 9 nt deletion status. Again, further study on Page serogroup B and Kume serovar B-1 isolates is required. As Page serogroup B strains are known not to be fully cross-protective, 13 the existence of additional Kume serovar B types seems likely.
Although the mPCR was proposed to be capable of recognizing the 3 Page serogroups, our work and that of an earlier study 6 has shown the shortcomings of the assay. A result in the mPCR of Page serogroup C can be accepted as correct, whereas a serogroup A or B result can be given with reference strains and isolates that are truly Page serogroup A, B, or C. The proposed PCR-RFLP, fully evaluated in our study, gives no effective improvement over the mPCR and is possibly too technically demanding for most diagnostic laboratories.
Region 2 sequence analysis, as undertaken in this work, seems to be helpful. However, more work is required on a larger number of field isolates that represent the full range of Kume serovars and/or Page serogroups as well as geographic diversity.
Footnotes
Acknowledgements
We thank Dr. W. Feng, China Institute of Veterinary Drug Control, and Dr. P. Zhang from our institute, for providing domestic isolates of Avibacterium paragallinarum.
Authors’ contributions
H Wang contributed to conception of the study; contributed to acquisition, analysis, and interpretation of data; and drafted the manuscript. H Sun contributed to conception of the study; contributed to acquisition and interpretation of data; critically revised the manuscript; and gave final approval. PJ Blackall contributed to analysis of data; critically revised the manuscript; and gave final approval. Z Zhang contributed to design of the study and contributed to analysis of data. H Zhou contributed to analysis and interpretation of data and critically revised the manuscript. F Xu contributed to interpretation of data. X Chen contributed to design of the study; contributed to acquisition of data; and drafted the manuscript. H Sun, PJ Blackall, and F Xu agreed to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
Pat Blackall, The University of Queensland, St. Lucia, Queensland, Australia.
b.
Taihe Biotechnology Co. Ltd., Beijing, China.
c.
DNAStar, version 7.1, DNASTAR Inc., Madison, WI.
d.
MEGA6.0, Center for Evolutionary Medicine and Informatics, Biodesign Institute, Arizona State University, Tempe, AZ.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants 31272558 and 30871867 from Natural Science Foundation of China, and National Key Research Program of China (2016YFD0500804).