
Abstract
Loop-mediated isothermal amplification (LAMP) is a sensitive method for DNA amplification. In the present report, the development of a single-tube, one-step, real-time accelerated reverse transcription (RT)-LAMP for the detection of Porcine teschovirus (PTV) is described. Six designed primers amplified target gene sequences successfully at constant temperature (65°C) within 1 hr, and the amplification results could be visualized directly by the naked eye. The sensitivity of the LAMP was 10 times higher than that of conventional polymerase chain reaction, and no cross-reactivity was found when the genomes of other common swine pathogens were subjected to the RT-LAMP system. When 43 clinical samples were tested by the RT-LAMP method, results indicated that the test is simple, rapid, accurate, and sensitive for the detection of PTV.
Porcine teschovirus (PTV; genus Teschovirus, family Picornaviridae) is the etiologic agent of teschovirus encephalomyelitis. A polioencephalomyelitis with high morbidity and mortality (known as Teschen disease) that occurred in Czechoslovakia in 1929 was the first report of porcine teschovirus infection. 14 This disease caused extensive losses in Europe and was spread to other continents during the 1940s and 1950s. Porcine teschovirus comprises at least 11 distinct serotypes. 19 Although infections with most PTV are often asymptomatic, virulent strains can give rise to a wide range of clinical signs, including polioencephalomyelitis, 6 infertility, 3 pneumonia, 9 enteric disease, 4 and dermal lesions. 4,18 Serotypes 1, 2, and 8 have so far been isolated in China. 2,5,15
Currently, techniques for the diagnosis of PTV infections include histological examination, viral isolation, serological testing, and molecular assays. A 2007 study described the detection of viral antigen by immunohistochemical (IHC) staining of formalin-fixed, paraffin-embedded tissue sections. 17 But IHC is often not suitable for large-scale testing because it is a labor-intensive process. Virus isolation, followed by observation of cytopathic effects in cell cultures, or in combination with immunological detection, can be used for PTV detection if suitable reference reagents (specific antiserum or monoclonal antibodies) are available. However, for successful viral isolation, samples of central nervous system must be collected at an early clinical stage of the disease. 8 Furthermore, virus isolation lacks the sensitivity 10 and is time-consuming, requiring at least 3–4 days. Molecular tests such as reverse transcription polymerase chain reaction (RT-PCR) and real-time RT-PCR assays have also been developed for diagnostic use. 7,12 Although PCR assays are sensitive methods, they require considerable operator skill and precision thermocycling equipment, which can be expensive and may not be available under field conditions. There is therefore a need to develop a simple, rapid, sensitive, and cost-effective method for PTV detection. A nucleic acid amplification method, termed loop-mediated isothermal amplification (LAMP), has been previously described. 11 In the present study, the development of a one-step reverse transcription (RT)-LAMP assay for PTV detection is reported.
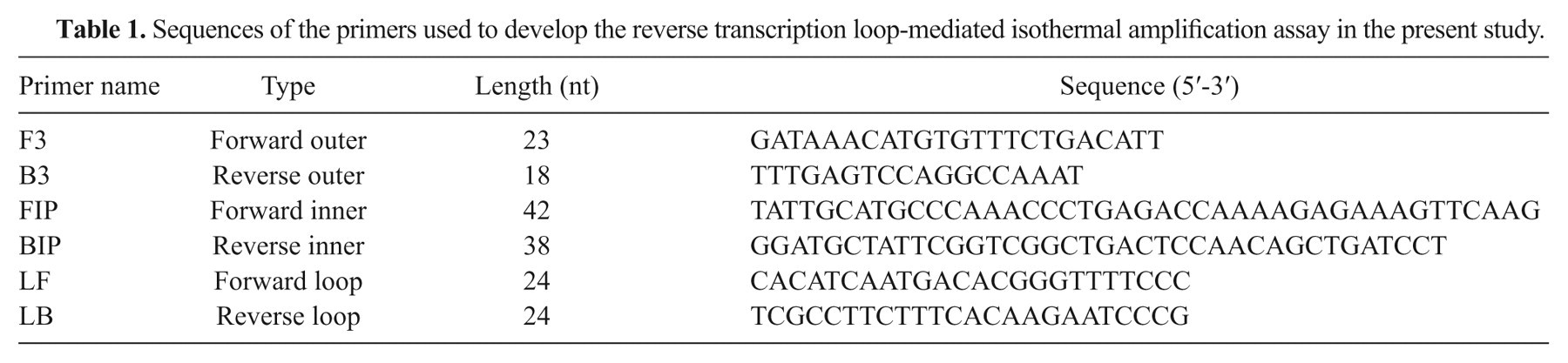
A set of RT-LAMP primers was designed targeting a region located in the RNA-dependent RNA polymerase (RdRp) gene, which was selected based on alignment analysis. The analysis involved 31 genomic sequences from 11 serotypes, retrieved from the GenBank database 15 and aligned by ClustalW a to identity the highly conserved region. The set of 6 primers composed of 2 outer (F3, B3), 2 inner (FIP, BIP), and 2 loop primers (F-loop, B-loop) that recognize distinct regions on the target site were designed by use of the LAMP primer design software PrimerExplorer version 4, b as described. 11 The sequences for each primer are shown in Table 1.
Sequences of the primers used to develop the reverse transcription loop-mediated isothermal amplification assay in the present study.
A previously described PTV strain isolated by the authors was used as reference strain to develop the RT-LAMP method. 15 Viral RNA was extracted from PTV-infected Instituto Biologico renal swine (IBRS-2) cell line with a commercial RNA extraction kit, c following the manufacturer’s instructions. The RT-LAMP reaction was performed using an RNA amplification kit d in accordance with the manufacturer’s protocol. Briefly, a 25-µl reaction mixture containing 40 pmol each of the primers FIP and BIP, 20 pmol each of the primers loop primer F and B, 5 pmol each of the primers F3 and B3, 12.5 µl of 2× reaction mixture, 5 µl of sample RNA, 1 µl of enzyme mixture, and 1 µl of fluorescent detection reagent (FDR), d was incubated at 65°C for 1 hr in a real-time turbidimeter, d followed by incubation at 80°C for 5 min to terminate the reaction. Results indicated that the positive signal could be detected in less than 30 min, but for optimum amplification, the reaction time of 60 min was selected. In order to facilitate field application of the RT-LAMP assay, reading of the assay was also carried out using the naked eye. In the presence of FDR, a positive reaction, resulting from successful amplification of PTV RNA was indicated by a color change from yellow to green.
To evaluate the specificity of the RT-LAMP, other common pathogens of swine prevalent in China were used, including Porcine reproductive and respiratory syndrome virus (PRRSV, CH1-a strain), highly pathogenic PRRSV (HuN4-112 strain), Classical swine fever virus (CSFV, C strain), Japanese encephalitis virus (JEV, SA14-14-2 strain), Transmissible gastroenteritis virus (TGEV, H strain), Swine influenza virus (SIV, A/Swine/Inner Mongolia/547/2001 (H3N2) strain), Porcine rotavirus (RV, JL94 strain), Porcine parvovirus (PPV, HLJ strain), Pseudorabies virus (PRV, Bartha-K61 strain), and Porcine circovirus (PCV, HLJ strain). These viruses were kept at National Key Laboratory of Veterinary Biotechnology of Harbin Veterinary Research Institute, the Chinese Academy of Agricultural Sciences. Viral RNA or DNA was extracted from these viruses as described 15,16 and then subjected to the RT-LAMP reaction. Both real-time monitoring and FDR staining showed no amplification of any of the other swine viruses, confirming the specificity of the assay for PTV.
The analytical sensitivity was determined by using a previously described method, 13 with minor modifications. In brief, a fragment of the RNA-dependent RNA polymerase gene containing the sequences between the 2 outer primers (F3 and B3) was amplified by RT-PCR with the upstream primer 5’-ACTCACTGCAGCTACC-3’ and downstream primer 5’-TTAGGCAACAGGCAG-3’. Amplification products were separated by electrophoresis and purified using gel extraction kits c and cloned into pMD18-T vector e according to the manufacturer’s instructions. The sequence-confirmed recombinant plasmid was linearized using a vector-specific restriction enzyme (SacI) and used as standard control. The concentration of the linearized plasmid preparation was determined with a commercial apparatus. f Template copy number was calculated as followed: copies/ml = 6.02 × 1023 (copies/mol) × concentration of nucleic acid (g/µl)/average molecular weight of nucleic acid (g/mol). A 10-fold dilution series of linearized plasmid was used as a template for LAMP. To compare the sensitivity of the LAMP assay and PCR, serial dilutions of the template were also subjected to conventional PCR with F3 and B3 primers under the following cycling conditions: initial denaturation at 95°C for 5 min, 30 cycles of template denaturation at 95°C for 30 sec, annealing at 51°C for 30 sec, extension at 72°C for 30 sec, and post extension at 72°C for 7 min. Results showed that the detection limit of LAMP was 1.5 × 102 copies whereas conventional PCR was 1.5 × 103 copies. Thus, LAMP was 10 times more sensitive than conventional PCR.
In order to assess the applicability of the RT-LAMP assay for detection of PTV RNA in clinical samples, 22 tissue samples (5 brains and 17 feces) from a previous experimental study were tested. Twenty-one field samples (3 brains and 18 feces) were also obtained from suspected PTV-infected pigs showing suggestive clinical signs, including pyrexia, diarrhea, and nervous disorders (difficulty in standing, recumbency, and hind limb paralysis), in a commercial pig farm. All specimens were tested by RT-LAMP and RT-PCR in parallel. For RT-PCR, reverse transcription was carried out under standard conditions using random 9-mers, and then PCR was performed in a total volume of 25 µl with previously published primers. 12 All the 22 experimental infection samples were positive for PTV RNA by RT-LAMP, whereas only 18 of the 22 were positive by RT-PCR (1 brain and 3 fecal samples were negative). In the case of field samples, all the samples negative by RT-LAMP were also negative by RT-PCR, but 2 fecal samples were positive by RT-LAMP and negative for RT-PCR (Table 2). These data indicated that RT-LAMP was more sensitive than conventional RT-PCR, especially in fecal samples, as determined in a previous study. 1 The RT-LAMP showed 83.7% agreement with RT-PCR, but it is concluded that RT-LAMP method is superior to RT-PCR.
Comparison between reverse transcription loop-mediated isothermal amplification (RT-LAMP) and reverse transcription polymerase chain reaction (RT-PCR) in detecting Porcine teschovirus RNA from clinical specimens.
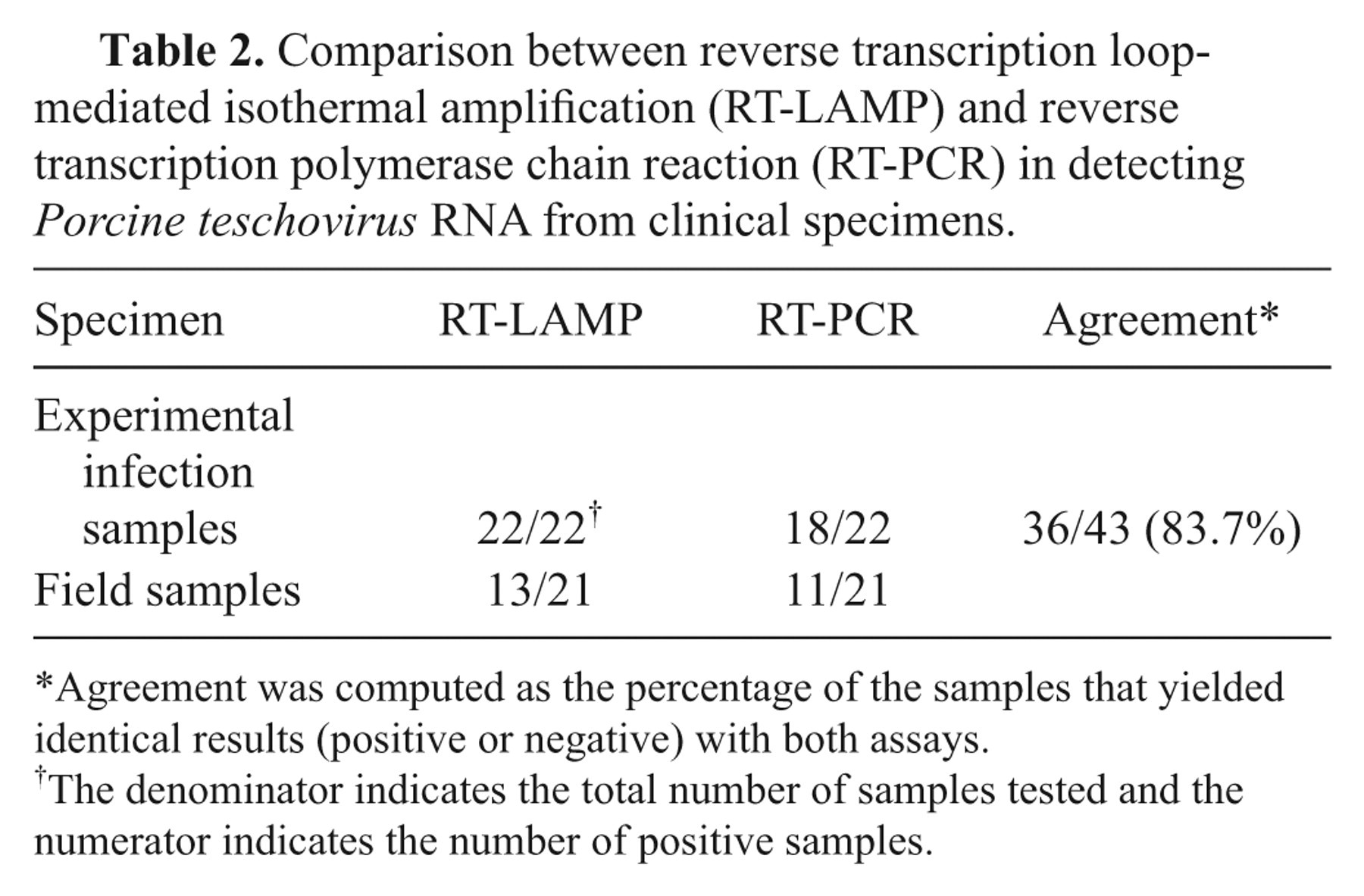
Agreement was computed as the percentage of the samples that yielded identical results (positive or negative) with both assays.
The denominator indicates the total number of samples tested and the numerator indicates the number of positive samples.
In conclusion, the RT-LAMP method developed in the present study exhibited good sensitivity and specificity for PTV genome detection. The reaction can be completed at constant temperature, thus only simple equipment, such as a heating block or water bath, is required, obviating the need for expensive automated thermal cyclers. The RT-LAMP results can be determined directly by the naked eye. This economic, easy-to-perform, and fast method would be useful for epidemiological surveillance of PTV and also for PTV infection diagnosis even under field conditions, such as pig farms.
Footnotes
Acknowledgements
The authors would like to thank Dr. Manabu Yamada of Japanese National Institute of Animal Health for his valuable discussion and Sheng-Zhi Zhang for his help with viral DNA extraction. The authors also thank the members of PRRS research group at Harbin Veterinary Research Institute of Chinese Academy of Agricultural Sciences for their assistance in collecting the clinical specimens.
a.
DNASTAR Inc., Madison, WI.
c.
Qiagen, Hilden, Germany.
d.
Eiken Chemical Co. Ltd., Tokyo, Japan.
e.
Takara Bio Inc., Dalian, China.
f.
Thermo Scientific, Shanghai, China.
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
The authors declared that they received no financial support for their research and/or authorship of this article.