
Abstract
Bovine viral diarrhea virus (BVDV) is classified into 2 genotypes, BVDV-1 and BVDV-2, each of which contains distinct subtypes with genetic and antigenic variation. To effectively control BVDV by vaccination, it is important to know which subtypes of the virus are circulating and how their prevalence is changing over time. Accordingly, the purpose of our study was to estimate the current prevalence and diversity of BVDV subtypes from persistently infected (PI) beef calves in the central United States. Phylogenetic analysis of the 5′-UTR (5′ untranslated region) for 119 virus strains revealed that a majority (82%) belonged to genotype 1b, and the remaining strains were distributed between genotypes 1a (9%) and 2 (8%); however, BVDV-2 subtypes could not be confidently resolved. Therefore, to better define the variability of U.S. BVDV isolates and further investigate the division of BVDV-2 isolates into subtypes, complete genome sequences were obtained for these isolates as well as representatives of BVDV-1a and -1b. Phylogenetic analyses of the complete coding sequence provided more conclusive genetic classification and revealed that U.S. BVDV-2 isolates belong to at least 3 distinct genetic groups that are statistically supported by both complete and individual coding gene analyses. These results show that a more complex set of BVDV-2 subtypes has been circulating in this region than was previously thought.
Introduction
Bovine viral diarrhea virus (BVDV; family Flaviviridae, genus Pestivirus) 7 is an important pathogen of cattle that causes respiratory disease, enteritis, and immune dysfunction. 2 BVDV can also cross the placenta and infect the fetus, resulting in abortion, congenital malformations, or birth of persistently infected (PI) calves. 2 Persistent infection develops when a susceptible dam is infected with a noncytopathic strain of BVDV and becomes viremic early in gestation, infecting the fetus prior to the maturation of its immune system. 25 Consequently, PI calves are born BVDV antibody negative, maintain a lifelong viremia, and continuously shed virus into the environment, making them the main reservoir for the virus and the leading source of transmission to susceptible cattle.2,9,12 As a result, herds with PI animals have higher production costs because of poor feed conversion, increased illness, and higher mortality rates. 17
BVDV has a single-stranded RNA genome that is ~12.3 kb and consists of a single open reading frame flanked by 5′ and 3′ untranslated regions (UTRs). Based on sequence comparison of the highly conserved 5′-UTR, BVDV isolates have been divided into 2 genotypes, BVDV-1 and BVDV-2,28,32 each of which contains distinct subtypes8,41 with genetic and antigenic 1 variation. Although both genotypes are diagnosed worldwide, the prevalence of subtypes varies by geographic region. Therefore, to effectively control BVDV by vaccination, it is important to know which subtypes of the virus are circulating and how their prevalence is changing over time. To date, 3 major subtypes reportedly circulate in the United States: BVDV-1a, -1b, and -2a9,10; however, the predominance of these subtypes has shifted from BVDV-1a to BVDV-1b over the past 20 years in BVDV strains characterized by diagnostic laboratories. 34 In addition, 3 U.S. isolates of BVDV were classified in 2014 as BVDV-2c based on complete genome sequencing. 19 There has also been a single case of BVDV-2b infection in the United States, 14 highlighting the need for continued surveillance to detect emerging viral subtypes. Given the importance of PI cattle in the transmission of BVDV, the purpose of our study was 2-fold: 1) compare the current regional prevalence of BVDV subtypes in PI beef calves with similar samples collected 10 years earlier and 2) investigate the genetic variation among these viruses with complete genome sequencing.
Materials and methods
Study population and sample collection
Calves sampled in our study were purchased by order buyers from at least 5 central states (Alabama, Mississippi, Missouri, Oklahoma, and Texas) and shipped to a southwest Kansas feedyard between August 2013 and April 2014. On feedyard arrival, all calves were tested for BVDV infection by Central States Testing (Sublette, KS) using a noncommercial antigen enzyme-linked immunosorbent assay (ELISA) on supernatants from ear notch specimens. Cattle that tested positive for BVDV were classified as PI and maintained in pens apart from the other animals. For this study, blood samples collected with EDTA as anticoagulant and ear notches were collected from 131 potential PI calves on April 30, 2014. Ear notches were sent to the University of Nebraska–Lincoln Veterinary Diagnostic Center for verification of BVDV PI status by immunohistochemistry (IHC). PI status was independently verified by reverse transcription (RT)-PCR using pestivirus-specific primers and cycling conditions listed below.
Cattle in this study were privately owned by a commercial feeding facility, and the owners and management approved the use of animals for this study. Animal welfare for the entire facility was in accordance with the National Cattlemen’s Beef Association’s Beef Quality Assurance Feedyard Welfare Assessment program (http://www.bqa.org/), and experimental protocols were approved by the University of Nebraska Institutional Animal Care and Use Committee (approval 1172).
Genetic typing of BVDV
BVDV was genotyped directly from noncultured clinical samples to avoid unnecessary handling and potential for contamination. Total RNA was extracted from 250 μL of plasma using a phenol and guanidine isothiocyanate reagent. a Pestivirus-specific primers (F: 5′-CATGCCCATAGTAGGAC-3′ and R: 5′-CCATGTGCCATGTACAG-3′ 13 ) were used to amplify a 283-bp fragment of the 5’-UTR. b Cyclic amplification reactions were carried out in a 25-μL reaction containing 2.5 mM MgCl2, 400 μM concentration of each dNTP, 0.4 μM concentration of each primer, and 1 μL of enzyme mix. b Cycling conditions were as follows: reverse transcription for 30 min at 50°C, inactivation of RT enzyme and activation of hot start polymerase for 15 min at 95°C followed by 40 cycles of 94°C for 30 s, 60°C for 60 s, and 72°C for 60 s. A final extension was carried out at 72°C for 7 min. Following amplification, the 283-bp fragment of the 5′-UTR was confirmed using agarose gel electrophoresis. These fragments were purified by exonuclease treatment, c according to the manufacturer’s specifications. DNA was then precipitated with ethanol and sequenced using the above pestivirus-specific primers. Sequencing was performed using dye terminator chemistry d and resolved on a capillary DNA analyzer. e
A neighbor-joining tree based on Hasegawa–Kishino–Yano (HKY) distance was produced using multiple sequence comparison by long-expectation (MUSCLE) algorithm for alignment of the 5′-UTR sequences using a commercial DNA analysis software package. f For construction of maximum likelihood trees, molecular evolutionary analysis software g was used to determine the most appropriate nucleotide substitution model using the best-fit model tool. The appropriate model was then used for phylogenetic inference according to the maximum likelihood algorithm using PhyML methods.f,15 Support for tree nodes was determined by 1,000 bootstrap analyses. Classification of subtypes was based on reference sequences from GenBank as reported previously.8,19,41
Generation of 19 full-length BVDV genomes
Nineteen full-length BVDV genomes were assembled de novo by sequencing virus strains directly from the plasma of infected calves as described previously. 43 This culture-free method was used to avoid the potential selection of a subpopulation of the infecting virus quasi-species when grown in cultured cells. Briefly, 100 ng of total plasma RNA were used as input material for an RNA library preparation kit. h Libraries were prepared as specified by the manufacturer’s protocol with 1 important modification: BVDV genomes lack a 3′ polyA tail, and thus the initial step of poly-A selection on oligo-dT beads was omitted. Libraries were sequenced on a desktop sequencer i with a 600-cycle kit (v3) to generate 2 × 300-bp paired-end reads.
Index adapters were removed from raw sequence reads using software,j,24 and trimmed reads were screened against a database,f,k to remove vector sequences. Given that virus strains were sequenced directly from the plasma of infected animals, host nucleic acids accounted for ≥98% of the reads. Therefore, assembly of viral genomic sequences was accomplished using template-assisted assembly where reference genomes (strain Singer, accession L32875; New York’93, accession AF502399; or Osloss, accession M96687) were used to discriminate viral from nonviral sequences in the dataset. Reads that mapped to the reference viral genome were then assembled de novo for each plasma sample. f
Phylogenetic and recombination analyses of full-length BVDV genomes
To compare isolates from this study with others from around the world, a neighbor-joining tree based on HKY distance was created from a MUSCLE alignment of the coding RNA sequence (CDS) of 19 full-length genomes generated in this study along with 75 strains available in GenBank. f The best nucleotide substitution model for maximum likelihood analyses was determined as described above. g The appropriate model was then used for phylogenetic inference according to the maximum likelihood algorithm using PhyML methods.f,15 The robustness of each tree was tested using 1,000 bootstrap analyses. GenBank accession numbers for strains used in these analyses are listed in Supplemental Table 1 (available online at http://vdi.sagepub.com/content/by/supplemental-data).
The MUSCLE alignment was also analyzed using a software package18,l to construct a neighbor-net phylogenetic network. The phi test for recombination 6 was used to evaluate the sequences for recombination. l Recombination was further evaluated using recombination detection software m using the default settings for a full exploratory recombination scan.22,23,27,30,35
Results
Genetic typing of BVDV
Of the 131 calves that tested positive for BVDV infection by antigen ELISA, 119 (91%) were verified as positive for persistent infection by IHC on skin (ear notch) specimens and by RT-PCR on plasma. The 12 animals not testing positive were removed from the study.
To determine the prevalence of BVDV genotypes in this population and assign each strain to a subgroup, phylogenetic analyses were performed on a fragment of the 5′-UTR with 2 commonly used methods, maximum likelihood (ML; Fig. 1A) and neighbor-joining (NJ; Supplemental Fig. 1, available online at http://vdi.sagepub.com/content/by/supplemental-data). These methods resulted in the identical classification of BVDV strains. Of the 119 confirmed PI calves, 110 (92%) were infected with BVDV-1. Of those, 99 (83.2%) were infected with BVDV-1b and 11 (9.2%) were infected with BVDV-1a. The remaining 9 (7.6%) were infected with BVDV-2 (Fig. 1A). No strains were classified as BVDV-2b; however, 2 strains, “83 TX” and “76 MO,” had higher pairwise sequence identity with BVDV-2c strains from Germany (98–99%) than prototypic U.S. BVDV-2a strains 890 (92%) and NewYork’93 (94%).
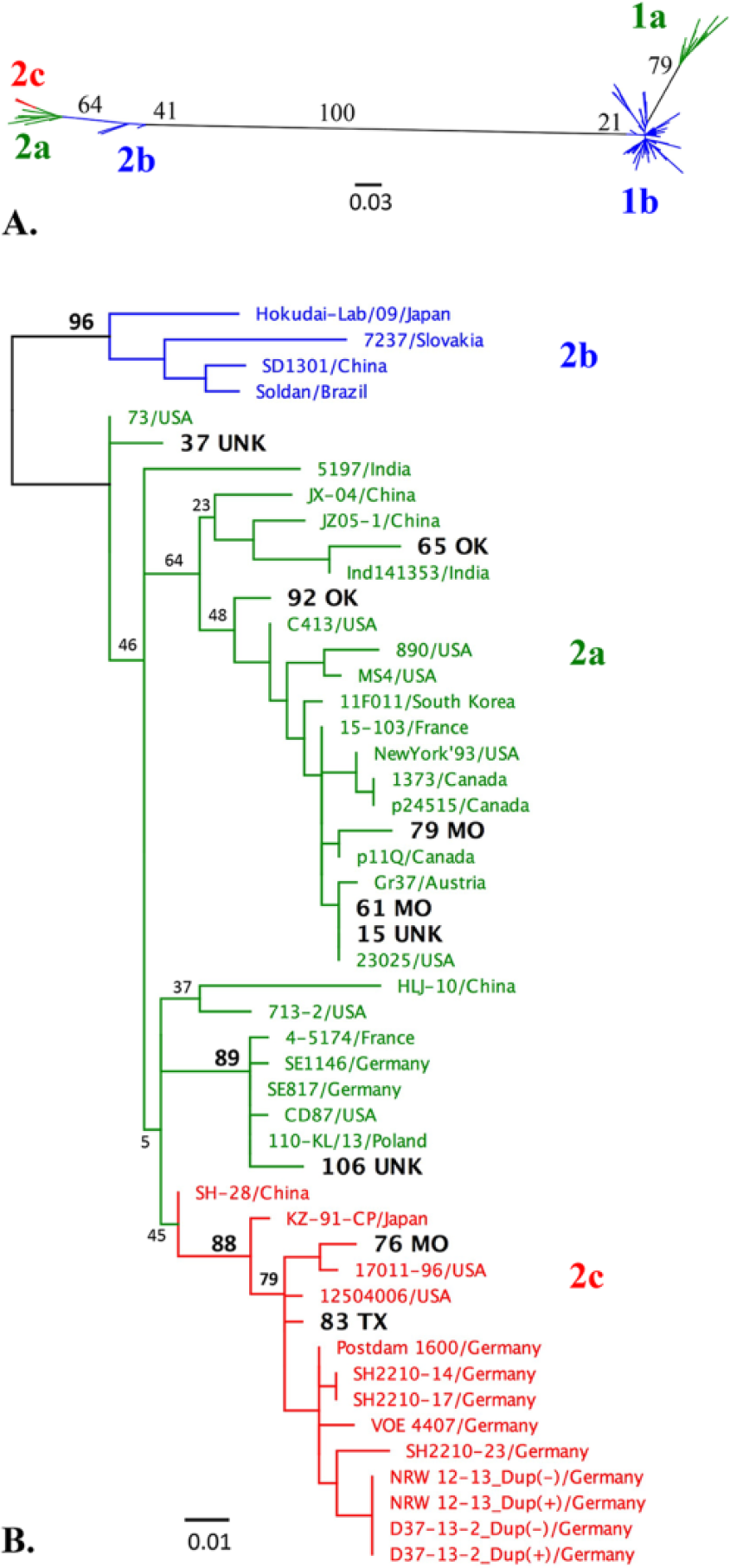
Maximum likelihood (ML) analyses of Bovine viral diarrhea virus (BVDV) 5′-UTR (untranslated region) sequences.
To better characterize BVDV-2 strains from our study, a basic local alignment search tool n was used to identify the most similar sequences in GenBank, and these strains were included together with additional reference BVDV-2 strains from around the world for phylogenetic analyses. Both ML (Fig. 1B) and NJ (data not shown) trees were generated. Separation of subtype 2b from the other BVDV-2 strains was strongly supported in both NJ and ML tree–based inferences. In contrast, the separation of BVDV subtypes 2a and 2c was not statistically supported by either method as evident from the low bootstrap values and variable strain placement between trees. Phylogenetic analysis and percentage of similarity determined by pairwise distances confirmed that 2 of the BVDV-2 strains from this study were most similar to BVDV-2c strains (bootstrap value >75%); and further revealed that one BVDV-2 strain from this study was most similar to a distinct subset of BVDV-2 isolates containing U.S. CD87 36 and German 2a strains 4 (bootstrap values >85%). However, given the overall low bootstrap values in the 5′-UTR NJ and ML trees, BVDV-2 strains could not be confidently subtyped.
BVDV prevalence by location
The prevalence of each genotype was also determined by the order buyer’s state of origin. The 119 PI calves were purchased at auction by order buyers from at least 5 central states; however, the precise geographic origin of each animal was not known. When analyzed by buyer’s state of origin, no significant difference was observed in the prevalence of BVDV subtypes (Table 1). Furthermore, there was no evidence to suggest geographic-related clustering of subtypes (Supplemental Fig. 1).
Prevalence of Bovine viral diarrhea virus (BVDV) subtypes in persistently infected cattle by order buyer location.*
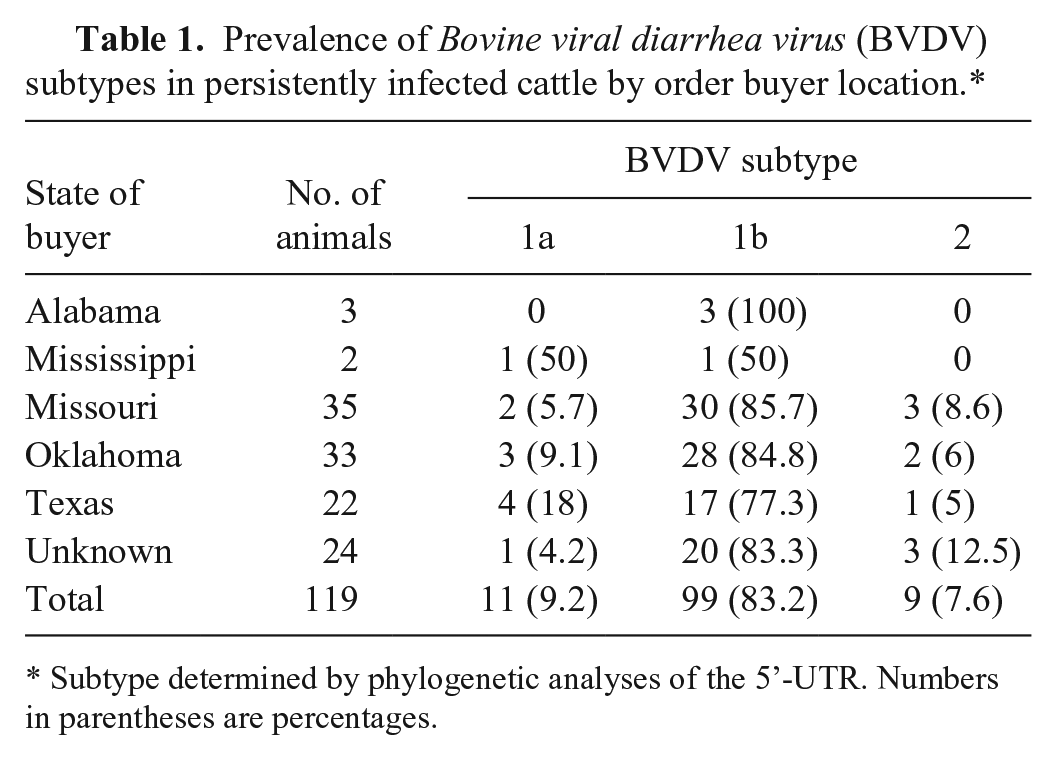
Subtype determined by phylogenetic analyses of the 5’-UTR. Numbers in parentheses are percentages.
Generation of 19 full-length BVDV genomes
To investigate the genetic variation among strains in our study and to increase the resolution of BVDV-2 subtyping, all 9 BVDV-2 strains, 2 BVDV-1a strains, and 8 BVDV-1b strains were selected from across the phylogenetic tree for complete genome sequencing (Table 2). Each of the 19 genomes assembled de novo into a single contig with an average genome length of 12,245 nucleotides (minimum 12,192; maximum 12,294) with mean read coverages from 33- to 1,059-fold.
De novo whole genome assemblies of 19 Bovine viral diarrhea virus (BVDV) strains.*
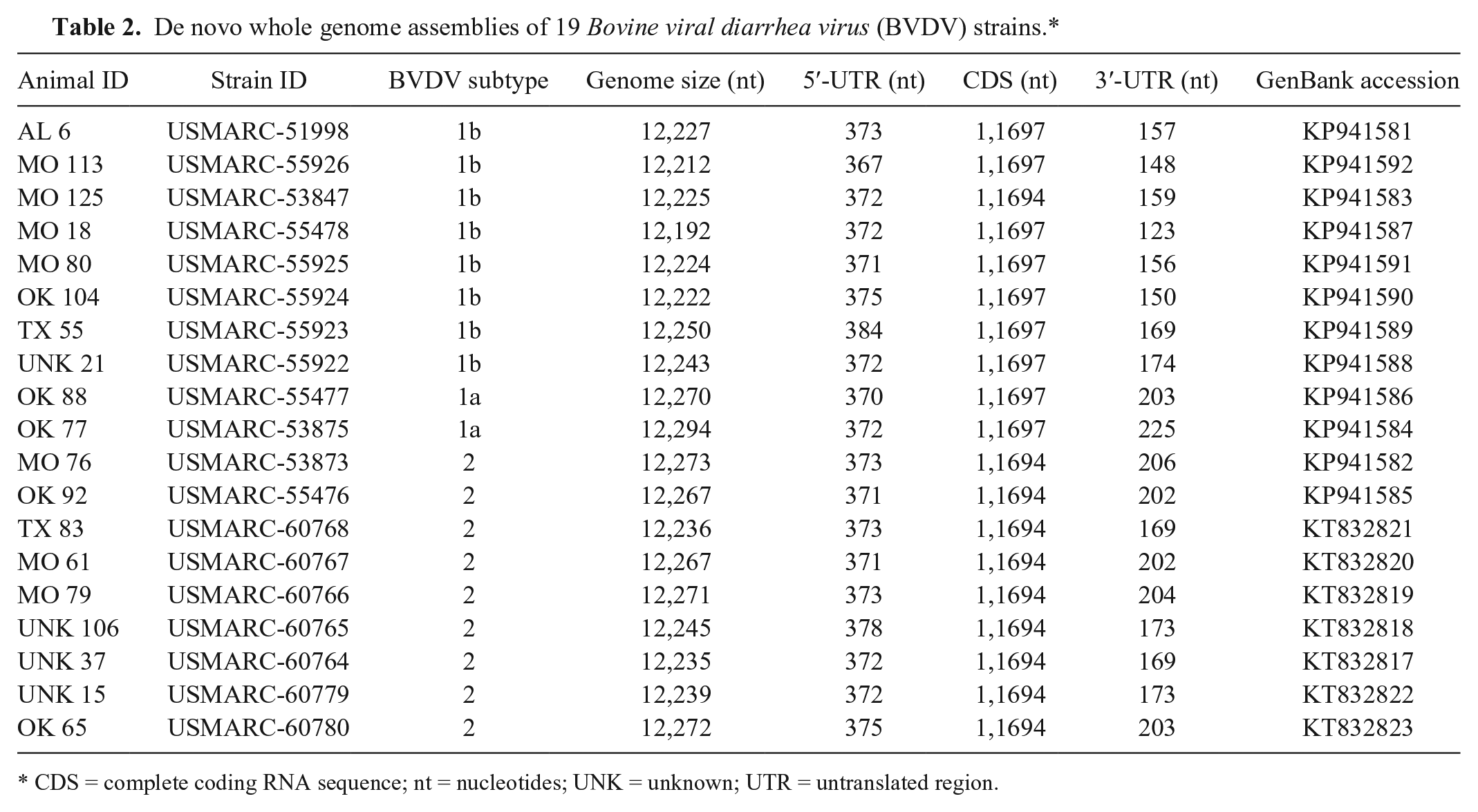
CDS = complete coding RNA sequence; nt = nucleotides; UNK = unknown; UTR = untranslated region.
The BVDV-1b strain USMARC-53847 from animal “125 MO” had a 3-nucleotide in-frame deletion in the E2 surface glycoprotein gene when compared to strains from this study as well as reference strains. No other deletions, insertions, gene duplications, or recombination events were detected in the coding sequences of the 19 genomes; however, 0–50 nucleotides were missing from the genome termini compared to reference strains.
Phylogenetic and recombination analyses of BVDV genomes
The CDSs of these 19 genomes were also compared to those of 75 full-length BVDV genomes isolated from around the world and available in GenBank. Phylogenetic analyses of the CDSs resulted in well-supported consensus trees, although there were some differences in the phylogenetic relationships inferred by the ML (Fig. 2) and NJ (Supplemental Fig. 2, available online at http://vdi.sagepub.com/content/by/supplemental-data) tree–based inferences. For BVDV-1, 100% concordance was observed between strain subtyping based on phylogenetic analyses of sequences from the 5′-UTR (Fig. 1) or the CDS (Fig. 2) for the 10 isolates from this study. Furthermore, the NJ and ML tree topology for BVDV-1 isolates was similar, although differences in the inferred relationships between the non–BVDV-1a or -1b isolates were observed.
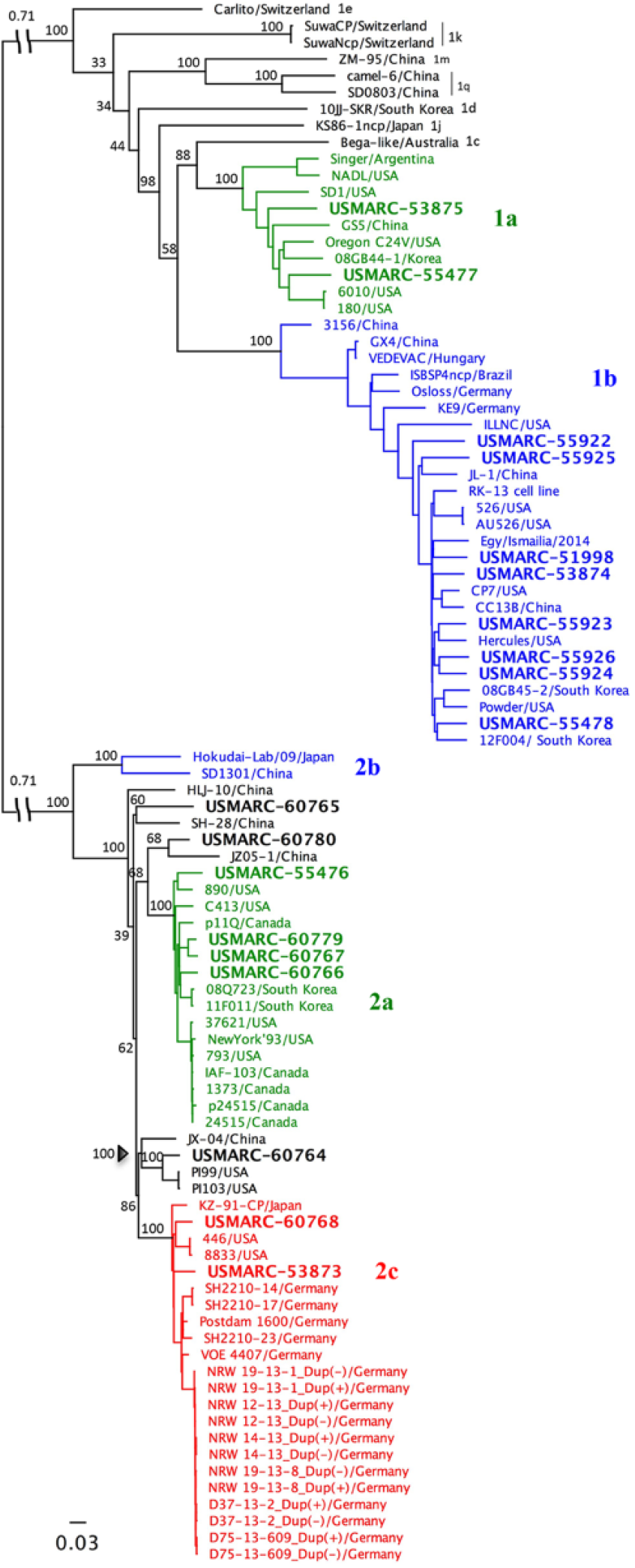
Mid-point rooted maximum likelihood (ML) tree of Bovine viral diarrhea virus (BVDV) complete coding sequences. A mid-point rooted ML tree was constructed on the basis of the complete coding RNA sequence of 19 strains from this study along with 75 full-length viral genomes available in GenBank (listed by their strain ID followed by country of origin). ML phylogeny construction was carried out using the general time reversible model of nucleotide substitution with a gamma shape parameter of 1.70 and a proportion of invariant sites equal to 0.41. The log likelihood of the inferred phylogeny is −190,930. Numbers indicate the percentage of 1,000 bootstrap replicates that support each group. The scale bar represents substitutions per site. Colors indicate BVDV subtypes.
For BVDV-2 stains, phylogenetic analysis of the complete CDS provided more conclusive genetic classification and revealed several distinct genetic clusters supported by high bootstrap values (Fig. 2). The first group contained the prototypic U.S. BVDV-2a strain 890 5 and 4 strains from our study (USMARC-60766, -60779, -60767, and -55476). The second group contained prototypic German BVDV-2c strains 19 and 2 strains from our study (USMARC-53873 and -60768). The third group contained BVDV-2b strains and no strains from our study. There were also several U.S. and Chinese strains as well as 3 strains from our study (USMARC-60764, -60765, and -60780) that were distinct from these previously defined clades (Fig. 2). JX-04/China, USMARC-60764, PI99/USA, and PI103/USA formed a group sister to the established BVDV-2c clade. This relationship was strongly supported in both NJ and ML trees. Similarly, USMARC-60780 and the recombinant strain JZ05-1/China 42 formed a group sister to the established BVDV-2a clade. Positioning of this group was identical in both NJ and ML trees. In addition, there were 3 single lineages (SH-28/China, USMARC-60765, and HLJ-10/China) that were distinct from the well-supported BVDV-2 clades. These strains had low bootstrap support and variable placement between NJ and ML trees. Therefore, the relationships among these strains could not be confidently resolved.
The low bootstrap values and variable placement of certain BVDV-2 strains between NJ and ML trees could be indicative of viral recombination.20,42 Therefore, sequences were tested for recombination using several algorithms within a recombination detection software package. m Strains 3156/China, JZ05-1/China, and ILLNC/USA were identified as recombinants as previously reported20,42; however some differences in parental strains were identified. 42 In particular, JZ05-1/China was previously found to have BVDV-2a 11F011/South Korea as the major parent and an unknown second strain as the minor parent. In contrast, this dataset found the major parent to be USMARC-60780. Neighbor-net phylogenetic networks were constructed to more accurately depict the phylogenetic relationships of viral recombinants (Fig. 3). No strains from our study tested positive for recombination.
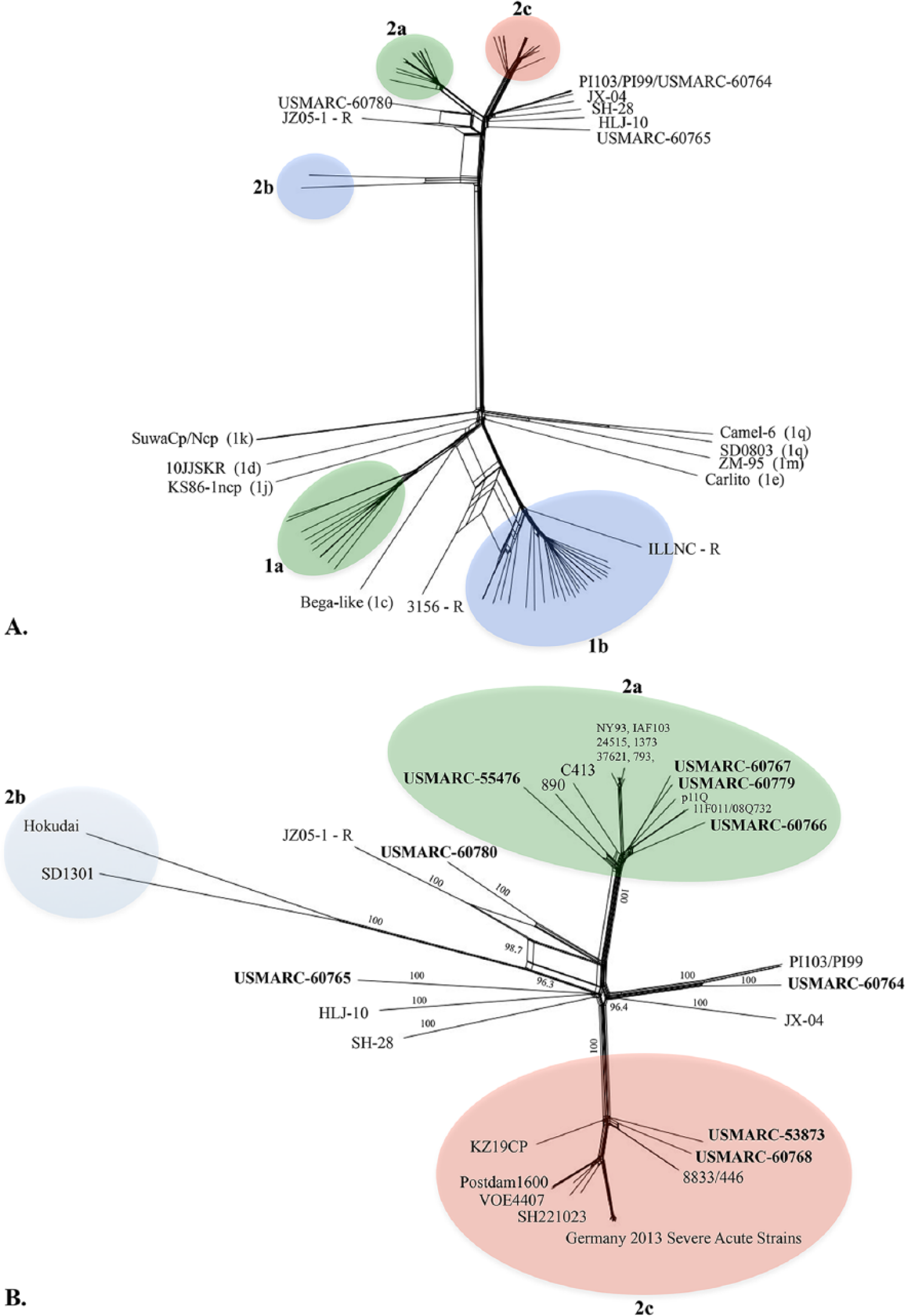
Neighbor-joining (NJ) phylogenetic network of Bovine viral diarrhea virus (BVDV) complete coding RNA sequences (CDSs).
The percentage of similarity of pairwise distances within and between BVDV groups was calculated. The nucleotide sequence divergence among virus isolates of the same group was 6% for BVDV-2a, 8% for BVDV-2c, and 11% for BVDV-2b. The ranges of sequence divergence between subgroups were 10% between BVDV-2a and -2c, and 16% between BVDV-2b and the other BVDV-2 clades. This is in contrast to BVDV-1 subtypes, which have a nucleotide sequence divergence of ≤16% among virus isolates of the same group and 17–23% divergence between subtypes (data not shown).
Individual gene analyses of BVDV-2 stains
To find a genomic region that provides a more stable, well-supported phylogeny than the 5′-UTR for the subtyping of BVDV-2 strains, phylogenetic relationships of individual genes were reconstructed by NJ and ML approaches. Similar tree topology and support were obtained using the 2 methods, and only the ML trees are shown (Fig. 4). Parameters used in ML phylogeny reconstruction are listed in Table 3. Several of the coding genes had strongly supported separation of BVDV-2a, -2b, and -2c clades with bootstrap values >95% (nodes marked by an asterisk [*] in Fig. 4). The strains that fall outside these clades were more weakly supported and had variable positions between individual gene trees, although they always fell outside the previously defined clades. Coding regions for NPro, E2, NS5A, and NS5B had the highest resolution and statistical support, with tree topologies most similar to the CDS (Fig. 4). NS2/3 also had high resolution and statistical support; however, recombinant strain JZ05-1/China was placed in the BVDV-2b clade for this genomic region.
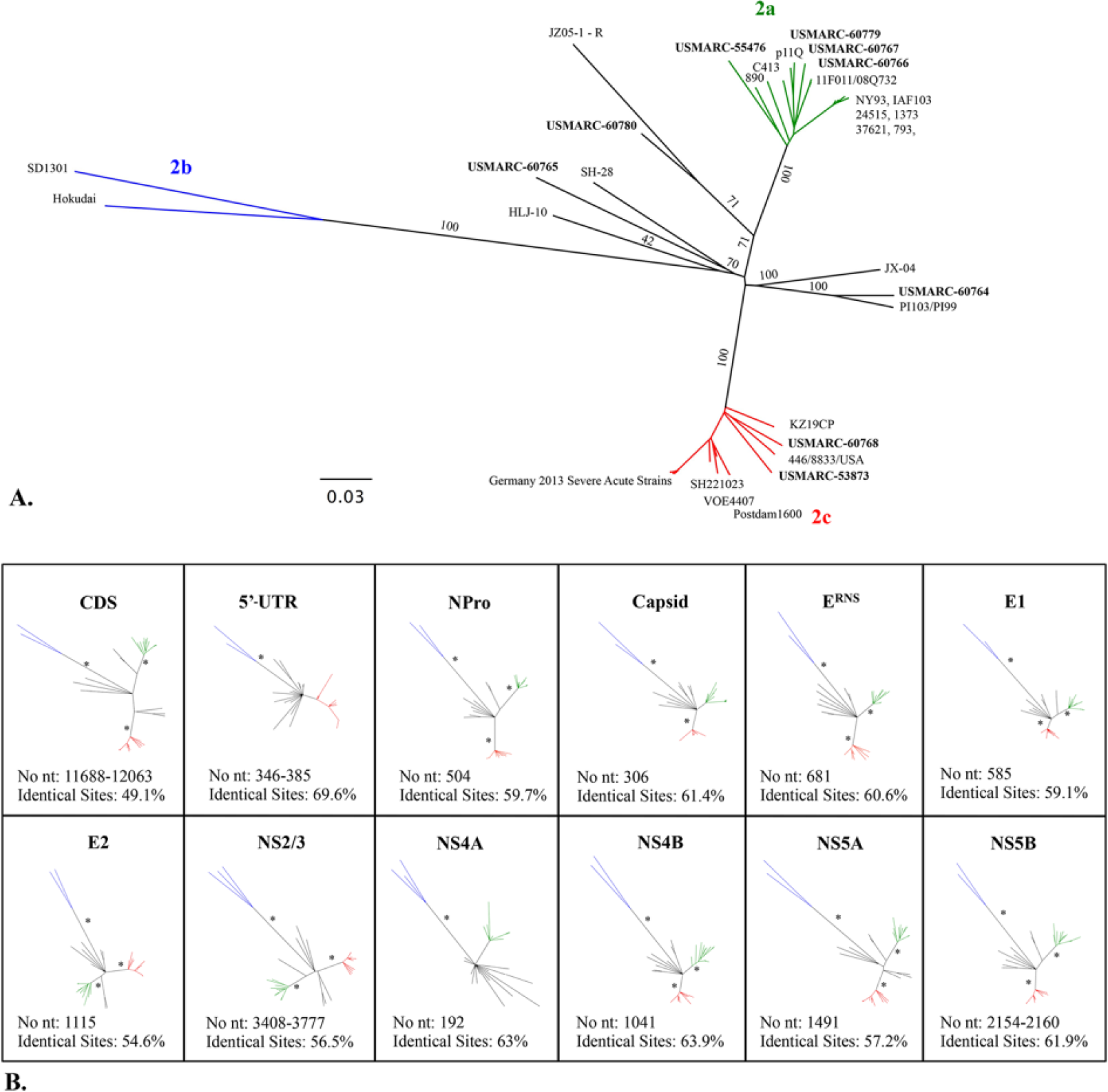
Individual gene phylogeny inferred by maximum likelihood (ML) analyses. Bovine viral diarrhea virus 2 (BVDV-2) phylogeny inferred by ML analyses of the complete coding RNA sequence (CDS;
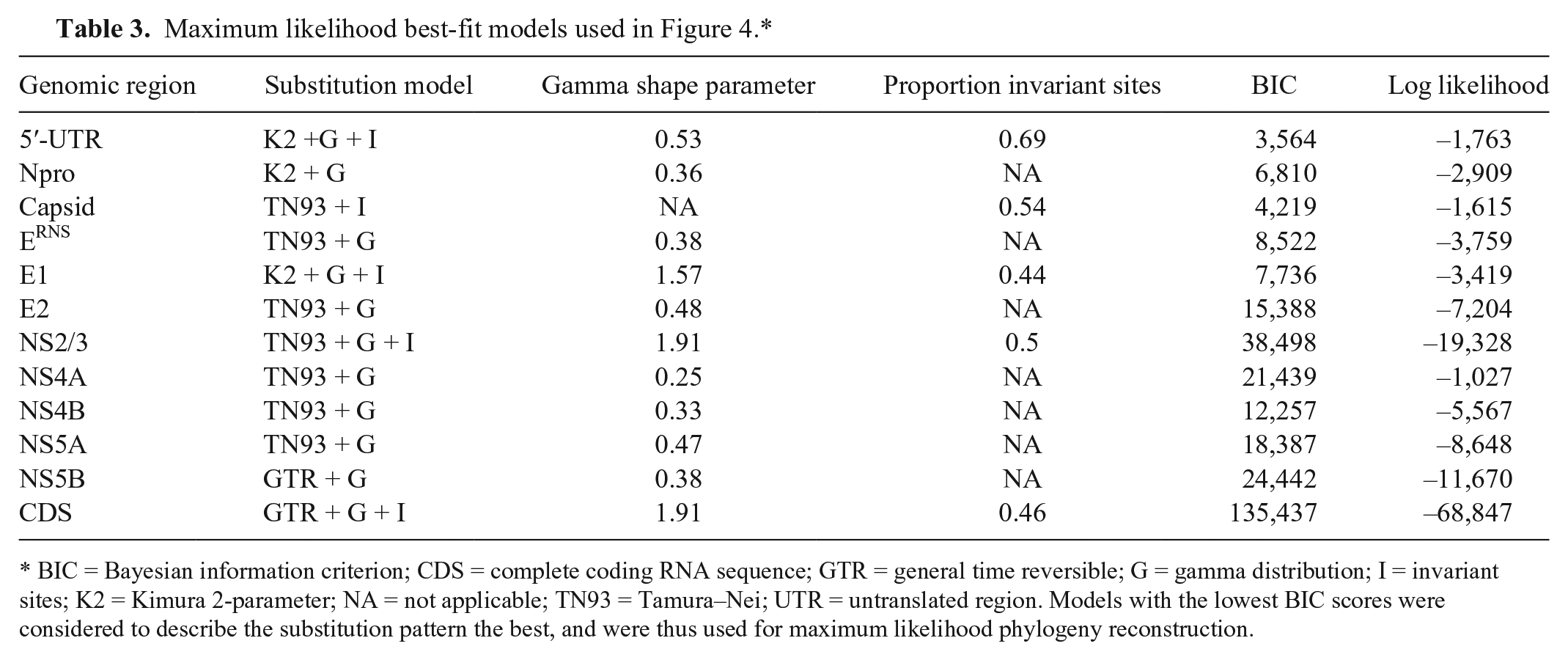
BIC = Bayesian information criterion; CDS = complete coding RNA sequence; GTR = general time reversible; G = gamma distribution; I = invariant sites; K2 = Kimura 2-parameter; NA = not applicable; TN93 = Tamura–Nei; UTR = untranslated region. Models with the lowest BIC scores were considered to describe the substitution pattern the best, and were thus used for maximum likelihood phylogeny reconstruction.
Discussion
The continuous and systematic characterization of BVDV strains circulating in the United States is needed by scientists, veterinarians, and ranchers to evaluate both prevention and control strategies, and to make informed management decisions. Therefore, the purpose of our study was to estimate the current prevalence and diversity of BVDV subtypes by sampling PI beef calves in the central United States Phylogenetic analyses of 119 viral strains from at least 5 central states revealed that BVDV-1b is the most common subtype infecting PI cattle in the region. These results are consistent with the high prevalence of BVDV-1b reported in previous studies.13,14,33,36 Interestingly, the genotype profile reported herein is comparable to that reported 10 years earlier at the same U.S. feedyard. 11 This suggests that the prevalence of subtypes in PI beef calves in the region has remained similar in the last decade despite potential changes in the source of animals, cattle management practices, vaccination protocols, or implementation of BVDV control programs during this time. Information regarding the use of vaccines in the herd of origin (breeding herd) for each animal was unavailable; thus, the impact of vaccination on subtype prevalence in this study is unknown.
Accurate genetic typing of viruses is important for the precise classification of new viral isolates, to understand what genetic diversity exists in a population, and to track virus evolution over time; however, uncertainty exists in BVDV classification caused by a lack of specific criteria for subtyping new virus isolates. 29 Previous phylogenetic studies on BVDV have been based on partial genomic sequences of the 5′-UTR or coding regions such as NPro, E2, or NS2/3. Of these, 5′-UTR is most commonly used and is ~380 nucleotides in length and contains both highly conserved regions as well as short variable regions.16,31 The conserved regions are important to allow detection of all pestivirus species; however, these regions contribute to the low bootstrap values observed in 5′-UTR phylogenetic trees.3,44 Despite this, it has been demonstrated that classification of BVDV-1 subtypes is consistent regardless of the genomic region used for analysis,3,37,39–41 with few exceptions resulting from homologous recombination.20,26,42 In contrast to what has been observed for BVDV-1 subtyping, our study revealed that BVDV-2 strains could not be confidently subtyped based on phylogenetic analyses of the 5′-UTR alone.
To better define the variability of U.S. BVDV isolates and further investigate the division of BVDV-2 isolates into subtypes, complete genome sequences were obtained by sequencing viral RNA from the plasma of infected calves. These full-length viral sequences were then compared with viral genomes from around the world. BVDV-2 subtypes were found to have much lower variability between subtypes (10–16%) than between BVDV-1 subtypes (20–24%). Nevertheless, phylogenetic analyses revealed 100% confidence in the subdivision of subtypes 2a, 2b, and 2c by NJ and ML methods. In addition, 3 strains from our study as well as strains previously classified as 2a were found to be distinct from these previously defined clades (Figs. 2–4). These strains had unstable placement in NJ and ML trees and could represent genetic intermediates; however, they tested negative for recombination, and thus are not genetic mosaics of the other subgroups. Additional full-length BVDV-2 sequences may help clarify the relationships between these strains.
Although analysis using the complete genome provides the highest resolution of the relationships among subtypes, it is not practical for large surveys. Therefore, single-gene analyses were carried out to determine which genomic segment most clearly resolves the relationship between subtypes and most accurately reflects the tree-based inferences using complete genome sequencing (Fig. 4). Although the relationships between the genetic intermediate strains were not fully resolved in the individual gene trees, many coding regions strongly supported the division of BVDV-2a, -2b, and -2c (Fig. 4). In addition, several regions further supported the division of the intermediate strains. However, recombinant strain JZ05-1/China placed in different clades depending on the genomic region analyzed, highlighting the problems with single-gene analysis for typing viruses capable of recombination.
In summary, the present study shows that the 5′-UTR can be used to segregate BVDV into major genotypes, but may be less suited for the segregation of BVDV-2 strains into subgroups. Coding regions provided more accurate subtyping of BVDV-2 strains; however, recombination events can lead to misclassification of strains typed by individual gene analyses. The clinical significance of BVDV-2 subtype division is currently unknown; further work is needed to determine the serologic relatedness of these subtypes. Regardless, the enhanced resolution of BVDV-2 subtypes provided by these analyses is critical for studying virus evolution and provides information for more accurate monitoring. This information is important to control programs and may influence the choice of BVDV strains to be used in vaccines.
Footnotes
Acknowledgements
We thank Brad Sharp, Sue Hauver, Bob Lee, Stacy Bierman, and Jacky Carnahan for technical support and Jan Watts for secretarial support. We also thank Steve Johnson for his assistance in the acquisition of samples for this study.
Authors’ contributions
AM Workman drafted the manuscript. All authors contributed to conception and design of the study; contributed to acquisition, analysis, and interpretation of data; critically revised the manuscript; gave final approval; and agreed to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Authors’ note
The use of product and company names is necessary to accurately report the methods and results; however, the United States Department of Agriculture (USDA) neither guarantees nor warrants the standard of the products. The use of names by the USDA implies no approval of the product to the exclusion of others that may also be suitable. The USDA is an equal opportunity provider and employer.
a.
TRIzol, Life Technologies, Grand Island, NY.
b.
OneStep RT-PCR kit, Qiagen Inc., Valencia, CA.
c.
Exo I, Life Technologies, Grand Island, NY.
d.
BigDye, PE Applied Biosystems, Foster City, CA.
e.
ABI 3730, PE Applied Biosystems, Foster City, CA.
f.
Geneious (version 8.0.5 21 ), Biomatters Inc., Newark, NJ.
g.
Molecular Evolutionary Genetics Analysis version 6 (MEGA6 version 6.06-mac 38 ), Arizona State University, Tempe, AZ.
h.
TruSeq RNA sample preparation kit, Illumina Inc., San Diego, CA.
i.
MiSeq, Illumina Inc., San Diego, CA.
j.
Cutadapt (version 1.9.1 22 ), Python Software Foundation, Beaverton, OR.
k.
UniVec_Core database, NCBI, Bethesda, MD.
l.
Splits Tree (version 4.13.1), University of Tübingen, Tübingen, Germany.
m.
Recombination detection program version 4, University of Cape Town, Cape Town, South Africa.
n.
BLAST, NCBI, Bethesda, MD.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: David Sjeklocha is employed by Cattle Empire LLC. Cattle Empire LLC did not have a role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for this research was provided by the United States Department of Agriculture, Agricultural Research Service (CRIS 3040-32000-031-00D).