
Abstract
Bovine viral diarrhea virus (BVDV) is an economically important pathogen of cattle. Two approved species are recognized, namely BVDV-1 and BVDV-2. To date, only 4 subgenotypes of BVDV-2 are known, and at least 11 distinct subgenotypes have been detected for BVDV-1. In a previous study, the genetic characteristics of 38 field isolates of BVDV from northern Italy were investigated, and all 38 isolates were classified as BVDV-1 and could be assigned to 5 different subgenotypes, namely BVDV-1b, BVDV-1d, BVDV-1e, BVDV-1h, and BVDV-1f. However, the circulation of BVDV-2 has been reported in Italy as well. The aim of the current study was to type 88 BVD viruses found throughout Italy. Genetic study was based on the 5′-UTR, supported by select comparison within the Npro coding region. Phylogenetic analysis showed that 5 isolates could be typed as BVDV-2a. The remaining 83 isolates were typed as BVDV-1 and were found to belong to 7 distinct subgenotypes, namely BVDV-1a (n = 8), BVDV-1b (n = 37), BVDV-1d (n = 3), BVDV-1e (n = 22), BVDV-1f (n = 4), BVDV-1g (n = 4), and BVDV-1h (n = 5). The majority of cattle farms in the current study were predominately infected by BVDV-1b and BVDV-1e isolates, whereas the other BVDV subgenotypes occurred only sporadically. The results also provided evidence for circulation of additional subgenotypes BVDV-1a and BVDV-1g. The occurrence of BVDV-2 was also reconfirmed.
Bovine viral diarrhea virus (BVDV) belongs to the genus Pestivirus, along with Classical swine fever virus and Border disease virus, in the family Flaviviridae. Presently, 2 species of BVDV are recognized, BVDV-1 and BVDV-2. Bovine viral diarrhea virus 1 infections occur worldwide and involve mainly respiratory, reproductive, and enteric organs, causing considerable economic losses in cattle production. Bovine viral diarrhea virus 2, discovered in North America 10,12 and also found worldwide, causes similar clinical signs as BVDV-1, except that infection with highly virulent isolates may lead to thrombocytopenia and fatal hemorrhagic syndrome.
The genome of BVDV consists of a single-stranded, positive-sense RNA approximately 12.3 kb in length. Genetic typing of BVDV has primarily been performed using sequences from the 5′-UTR, Npro, and envelope 2 regions. 1,2,9,12,15,18 Sequence analysis of 5′-UTR can distinguish between BVDV-1 and BVDV-2 genotypes; moreover, the same region can subdivide BVDV-1 into at least 11 subgenotypes, 18 indicating considerable genetic diversity within this pestivirus genotype. Cluster analysis of combined nucleotide sequences from the 5′-UTR and Npro regions leads to a higher statistical support by bootstrap analysis, although the grouping of isolates in 5′-UTR and Npro is the same. 18 Genetic typing of BVDV-2 isolates has not been as extensive, probably because of the smaller collections of virus isolates available for this type of analysis. To date, only 2–4 genetic groups of BVDV-2 have been described, 6,7 depending on whether the classification is based on nucleic acid sequence or on secondary palindromic structure. Studies on the prevalence of BVDV in Italy have provided evidence of the circulation of 5 BVDV-1 subgenotypes. 4 Bovine viral diarrhea virus 2 circulation has been reported in cattle as well. 8 In southern Italy, BVDV-2 has been circulating since the 1990s in sheep and goats. 11 To better define the genetic pattern within BVDV Italian isolates, a broad range of BVDV isolates collected from sick or healthy and persistently infected cattle from farms located all over Italy were studied. Genetic study was based on the 5′-UTR, supported by selected comparison within the Npro coding region.
Eighty-eight BVDV strains were collected, mainly during a period of 8 years (2000–2007), from 12 out of 20 Italian regions. The viruses were from cattle (n = 81), buffalo (n = 3), and sheep (n = 4). Fifty-one isolates originated from farms located in northern Italy, which has the highest cattle population density in the country.
The samples came from individual dead animals (n = 31), aborted fetuses (n = 2), and persistently infected animals (n = 55). Samples were sent to laboratories for routine testing because BVDV infection was suspected. All but 3 submitted samples (e.g., serum, blood, and tissue homogenates) were tested without virus isolation to avoid possible contamination of samples during cell culture by a pestivirus from fetal calf serum or the cell culture itself. Origins and characteristics of samples are reported in Table 1.
Total RNA was extracted using TRIzol reagent, a according to the manufacturer's recommendation. Synthesis of complementary DNA was carried out using random primers. b The genomic region encoding the highly conserved 5′-UTR of the pestivirus genome was amplified using primers 324 and 326 flanking a 288-base pair (bp) DNA fragment. 19 For 18 selected viruses, the genomic region encoding autoprotease Npro was amplified using a nested polymerase chain reaction (PCR). In the first PCR, the outer primers OI 100 and OL 1400R 2,3 were used, and reverse transcription PCR (RT-PCR) was done using the SuperScript III One-Step RT-PCR, a according to the manufacturer's recommendation. In the second PCR, the first-round PCR product was amplified with primers BD1/BD3 flanking a 428-bp fragment. 18 Where multiple fragments were obtained, a 428-bp fragment was cut from the gel and purified using the QIAquick gel extraction kit. c
Description of 88 Bovine viral diarrhea virus isolates characterized in the current study.*
Continued.
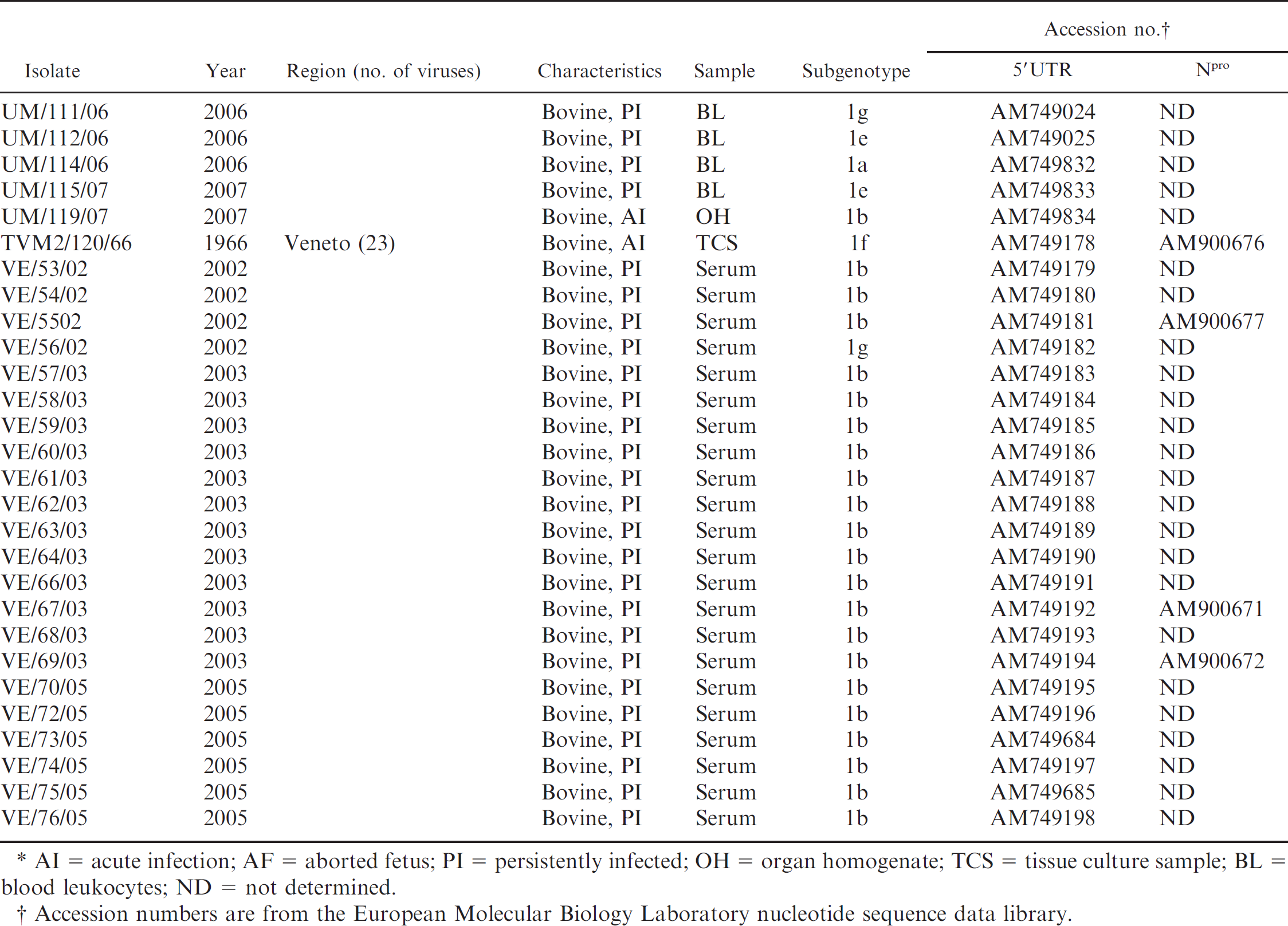
AI = acute infection; AF = aborted fetus; PI = persistently infected; OH = organ homogenate; TCS = tissue culture sample; BL = blood leukocytes; ND = not determined.
Accession numbers are from the European Molecular Biology Laboratory nucleotide sequence data library.
Nucleotide sequences were determined by cycle sequencing using the Big Dye Terminator v1.1 Cycle Sequencing kit d and an ABI PRISM sequencing device. d Nucleotide sequences were aligned using the ClustalX version 1.83 analysis program. 14 Phylogenetic trees were calculated using the PHYLIP version 3.66 program package based on the neighbor-joining algorithm method. 13 The robustness of the phylogenetic analysis and significance of branch order were determined by bootstrapping method carried out on 10,000 replicates using PHYLIP program SEQ-BOOT. 5 Sequence data from the current report have been deposited in the European Molecular Biology Laboratory nucleotide sequence data library (see accession numbers in Table 1).
All 88 viruses were sequenced in the 5′-UTR. Genetic typing revealed that 83 isolates were BVDV-1, and the topology of the tree (Fig. 1) indicates that they belonged to 7 distinct subgenotypes, namely BVDV-1a (n = 8), BVDV-1b (n = 37), BVDV-1d (n = 3), BVDV-1e (n = 22), BVDV-1f (n = 4), BVDV-1g (n = 4), and BVDV-1h (n = 5). To confirm the grouping found in the 5′-UTR, the Npro region of 18 viruses representative of each subgenotype and selected based on their bootstrap value were analyzed. As a result of the small number of BVDV-2 isolates, only BVDV-1 isolates were further compared. The resulting phylogenetic tree (Fig. 2) shows that these viruses were clustered in the same phylogenetic branches as the tree based on the 5′-UTR, with similar bootstrap values. Nonbovine isolates were all typed as BVDV-1. No relationship was observed between the geographic origins of the viruses and their phylogenetic clustering, with the exception of BVDV-1b, which is clearly predominant in northeastern Italy.
Five viruses that originated from the regions of Lombardia and Basilicata were typed as BVDV-2 and could be grouped into the subgenotype BVDV-2a. At the subgroup level, pairwise similarity and cluster analysis provided a clear-cut assignation to 7 distinct subgenotypes of 83 isolates typed as BVDV-1. Most cattle farms were infected by the predominant BVDV-1b and BVDV-1e isolates; other subgenotypes occurred only sporadically.
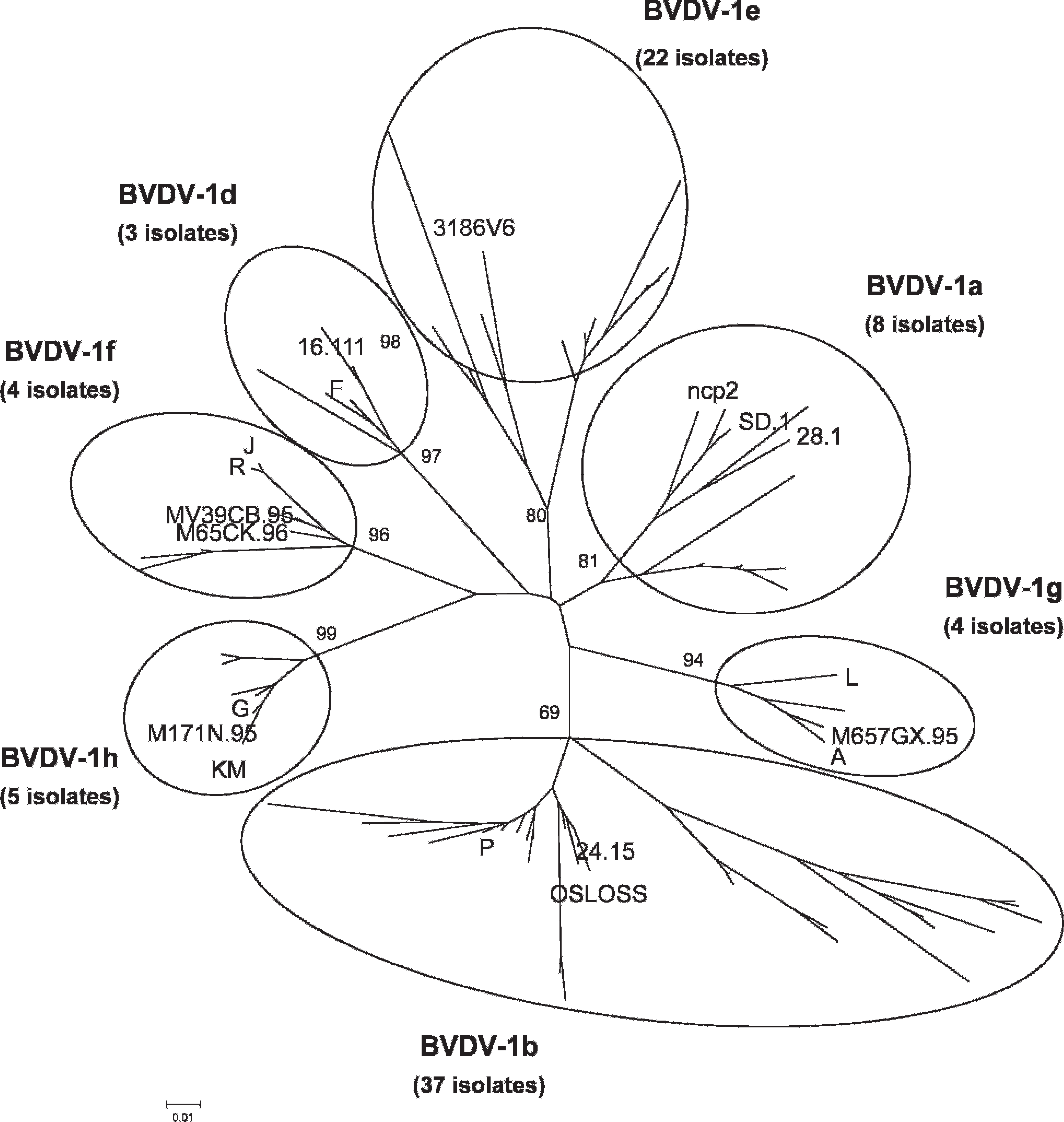
Phylogenetic tree showing the genetic relationship between Bovine viral diarrhea virus 1 isolates based on analysis of 238 nucleotides derived from the 5′-UTR. The tree contains all the isolates sequenced in this study plus 19 reference strains. It was prepared using the neighbor-joining algorithm with the Kimura 2-parameters. Numbers at the phylogenetic branches indicate the percentage of 10,000 bootstrap replicates that support each group. The following sequences for reference strains were acquired from GenBank: 16.111 (AF298056), 24.15 (AF298060), 28.1 (AF298061), 3186V6 (AF298062), A (298064), F (AF298065), G (AF298066), J (AF298067), KM (AF298068), L (AF298069), M171N.95 (U97431), M657GX.95 (U97455), M65CK.96 (U97456), MV39CB.95 (U97465), ncp2 (AY443027), OSLOSS (M96687), P (AF298070), R (AF298071), and SD.1 (M96751). Bar = number of substitutions per site.
This finding is consistent with previous observations on the predominance of these phylogenetic groups in several European countries. 16 The results also provided evidence for circulation of BVDV-1a and BVDV-1g subgenotypes, which have never been reported in Italy. As described previously, 17,18 for all the viruses genetically typed using combined 5′-UTR and Npro sequences, it was found that this comparison led to identical grouping. Five field viruses were typed as BVDV-2 and were clustered into the genotype BVDV-2a. They were isolated from both sick and apparently health animals. It is noteworthy that in Italy little is done to prevent the spread of BVDV; nevertheless, the emergence of BVDV-2 in the field is very uncommon.
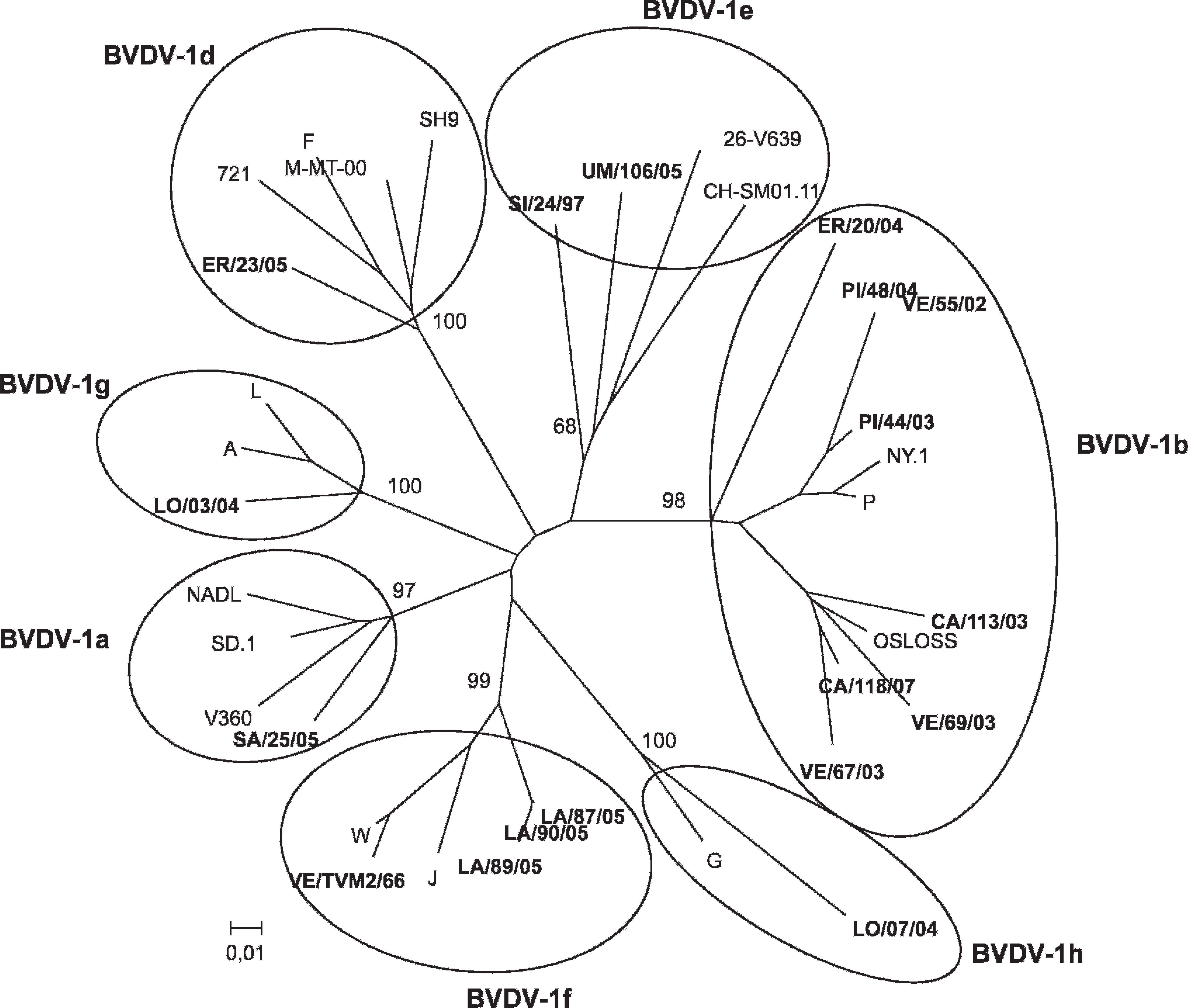
Phylogenetic tree showing the genetic relationship between Bovine viral diarrhea virus 1 isolates based on analysis of 359 nucleotides derived from the Npro gene. The tree contains 18 selected isolates (shown in bold type) representative of those analyzed using sequences from the 5′-UTR plus 24 reference strains. It was prepared using the neighbor-joining algorithm with the Kimura 2-parameters. Numbers at the phylogenetic branches indicate the percentage of 10,000 bootstrap replicates that support each group. The following sequences for reference strains were acquired from GenBank: NADL (M31182.1), SD.1 (M96751.1), V360 (AF144471.1), OSLOSS (M96687), NY.1 (AF145363.1), P (AF287288.1), SH9 (AF144473.1), M-MT-00 (AY323891.1), F (AF287284), 721 (AF144463), 26-V639 (AF287281), CH-SM01.11 (AY895000.1), A (AF287283), G (AF287285), J (AF287286), L (AF287287), and W (AF287290). Bar = number of substitutions per site.
Interestingly, the viruses isolated from buffalo and typed as BVDV-1b came from fetuses that were in the seventh month of gestation and from the maternal blood in the absence of anti-BVDV antibodies, indicating that this virus may be responsible for immunotolerance and concomitant persistent infection in adult animals. Thus, BVDV could act in the buffalo through the same pathogenic mechanisms that are well known in cattle.
In summary, the results presented in this work revealed a high BVDV genetic heterogeneity in Italy. This is the result of the absence of any BVDV systematic control measures. Indeed, in Italy, there is no BVDV national control program, and the management practices, such as cattle trade and movement, expose cattle herds to a high risk of introduction of BVDV infection as well as of new genetic variants of BVDV as a consequence of a high diversity of BVDV. This high genetic diversity leads to the question of whether BVDV vaccines, which have historically contained only BVDV-1a and BVDV-1b subgenotypes, can protect against infection with all of the highly diverse BVDV-1 isolates. According to the present study, that cannot be stated, and more definitive cross-protection studies are needed to address the importance of epitopic diversity and to determine whether future vaccines should be a mixture of several subgenotypes.
Acknowledgements. The authors thank Dr. G. Cardeti (IZS Lazio e Toscana, Roma), Dr. N. Cavaliere (IZS Puglia e Basilicata, Foggia), Drs. P. Cordioli and M. Luini (IZS Lombardia e Emilia, Brescia), Dr. G. Galiero (IZS del Mezzogiorno, Portici), Dr. A. Guercio (IZS Sicilia, Palermo), Dr. L. Masoero (IZS Piemonte, Liguria e Valle d'Aosta, Torino), Dr. S. Nardelli (IZS delle Venezie, Padova), Dr. G. Puggioni (IZS Sardegna, Sassari), and Professor S. Rosati (Università degli studi di Torino, Facoltà di Medicina Veterinaria) for providing BVDV isolates and epidemiological information. This work was supported by the Ministry of Health, RC IZSUM 05/2004 (D. Lgs 502/92, art. 12; D. Lgs 229/99, art. 12/bis).
Footnotes
a.
Invitrogen Corp., Carlsbad, CA.
b.
Amersham Biosciences Corp., Piscataway, NJ.
c.
Qiagen GmBH, Hilden, Germany.
d.
Applied Biosystems, Foster City, CA.