
Abstract
The objective of our study was to characterize the Mycoplasma hyopneumoniae genetic diversity within a swine operation comingling weaned pigs. Bronchial swabs and tracheal aspirates were collected from 3 nursery-to-finish farms. During the finishing production stages, samples were obtained from mortalities and from live coughing pigs in rooms where mortality was not observed. A total of 105 samples were examined by a M. hyopneumoniae real-time polymerase chain reaction and subjected to genetic typing using a multilocus variable number tandem repeat analysis (MLVA) assay. The MLVA was used to identify genetic variants based on the number of repeats in 2 variable number tandem repeats loci, namely P97 and P146, thought to mediate adherence of M. hyopneumoniae to swine cilia. Four distinguishable M. hyopneumoniae variants were identified: MVLA variants 9-15, 11-21, 9-21, and 7-15. Variant 9-15 was the most prevalent, observed in 79% of rooms, and detected on all 3 farms. Variant 11-21 was present in 37% of the rooms on 2 of the 3 farms. Only one 9-21 variant was identified in 1 farm, and all samples of variant 7-15 were recovered from another farm. Based on the low prevalence and limited geographic distribution of the last 2 variants, it is hypothesized that they might be the result of in-situ recombination. All variants detected in this investigation appeared to belong to 3 clusters. Overall, a limited number of variants and clusters were identified in a system that comingles pigs from different sources, suggesting limited M. hyopneumoniae genetic variation within commercial swine production environments.
Enzootic pneumonia, caused by Mycoplasma hyopneumoniae, continues to be a significant respiratory disease problem in swine production worldwide. 7 Enzootic pneumonia leads to decreased average daily gain and increased number of days to market weight, 4 and places a burden on animal welfare. Historically, M. hyopneumoniae infections have been controlled using various strategies, such as the adoption of optimal management practices and the use of vaccination, medication, and/or pathogen elimination. 7 Improvements in M. hyopneumoniae disease control include the implementation of novel management practices such as moving pigs in all-in/all-out fashion, and using multi-farm production, 3 the approval of new vaccines and antibiotics, the application of vaccination protocols at younger ages—including pig vaccination in the farrowing house, 5 and the use of antimicrobial treatments targeting critical times when transmission events are most likely to occur. 8
Strains of M. hyopneumoniae have been shown to express differences in virulence at the genome and proteome levels.1,9,10 These differences have led to a concern among swine veterinarians that wide genetic variation may influence M. hyopneumoniae infections and disease control in the field. The extent of M. hyopneumoniae genetic variability within large production systems, a common feature in the United States, has not been investigated and deserves further research. Therefore, the objective of our field investigation was to characterize the genetic diversity of M. hyopneumoniae variants within a swine production system.
Three nursery-to-finish farms (A–C) from a swine operation in the United States that raises half a million pigs per year were selected for this investigation. All 3 farms sourced pigs from 4–8 sow farms. Farm A received weaned pigs from 8 sow farms, farm B from 6 sow farms, and farm C from 4 sow farms. The rooms at each of the farms had weaned pigs from 1 or more of the sow farms (Fig. 1). All sow farms received breeding stock from a single pig multiplier. Semen was received from 1 in-system boar stud. Each farm enrolled in the study was National Pork Board Pork Quality Assurance Plus certified, and pigs were cared for following industry-approved standards. Farm A consisted of 13 rooms, whereas farms B and C consisted of 5 and 4 rooms, respectively, and each room housed ~1,000 pigs. Farm A was located 1 km apart from farm B, and farm C was separated by 20 km from farms A and B. Within each farm, all rooms were separated 15 m from each other and were either naturally or tunnel ventilated. At each farm, the same set of caregivers took care of the pigs and moved from room to room to carry out their daily chores without changing coveralls or using disinfection foot baths between rooms. A diagram of the herd and the 3 farms is shown in Figure 1.
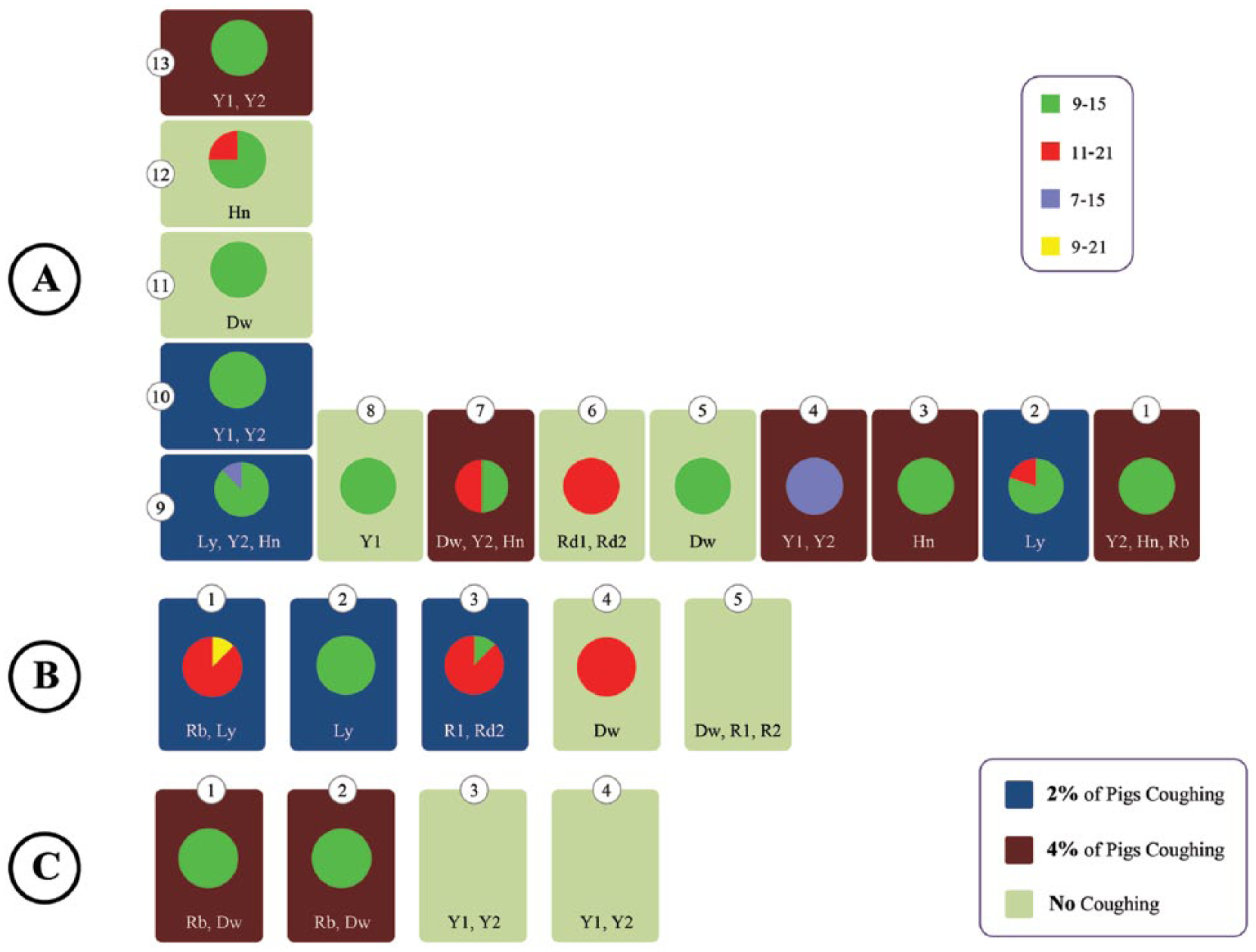
Mycoplasma hyopneumoniae genetic relationships at farms A–C. At each farm, each room is represented showing the room number, the code for the sow farm(s) from which the pigs originated, and a pie chart showing the distribution of M. hyopneumoniae multilocus variable number variants. The figure also indicates which rooms did not contain any coughing pigs and rooms that contained some pens with ~2% or 4% of pigs with dry coughing.
Pigs in the production system were negative for Porcine reproductive and respiratory syndrome virus and positive for M. hyopneumoniae under a control program. Prior to the initiation of our study, the production system veterinarian reported that pigs were coughing and experiencing sudden deaths caused by gastric ulcers at the finishing stages. Sows were vaccinated against M. hyopneumoniae 8 weeks prior to farrowing, and piglets were vaccinated at weaning age (~21 days of age). Thus, all pigs and sows in the system received a single full dose of a M. hyopneumoniae bacterin. a
For M. hyopneumoniae sample collection, pigs of 4.5 months of age were monitored for a period of 4 weeks during the summer months. Once during this time period a veterinarian visited the farm and inspected each room, and recorded the percentage of pigs coughing per pen on all 3 farms. First, prior to scoring each pen, the pigs were made to stand up when the veterinarian walked the length of the room. This was done to ensure that most pigs were active during the coughing evaluation time. The proportion of pigs exhibiting a dry cough during 1 min of observation was recorded per pen in each room. The number of coughing pigs was divided by the total number of pigs in the pen. A full autopsy was performed on all pigs that died during the 4-week collection period, and macroscopic postmortem findings were recorded.
Bronchial swabsb,6 were collected from on-farm mortalities, regardless of cause of death, as the overall goal was to obtain samples from all rooms and farms. As well, 28 tracheal aspirates were collected from rooms exhibiting dry cough but without mortalities. In brief, pigs were manually restrained with a cable snare while a flexible plastic tube of 1 cm in diameter was introduced through the mouth in order to reach the trachea. A source of artificial light was used to properly locate the opening of the larynx. Tracheal aspirates were collected with tubing cut to the approximate length of nose to tracheal bifurcation plus 10 cm and by applying gentle suction until resistance was met. Once retracted, typically 5–10 mm of mucus was present in the end of the tube (mucus was not weighed during this procedure). The mucus was then recovered from the tubing via swab b and submitted to the diagnostic laboratory c for testing.
To determine M. hyopneumoniae genetic variation, bronchial and tracheal aspirates were processed for DNA extraction and tested at the diagnostic laboratory c using a M. hyopneumoniae species-specific quantitative real-time polymerase chain reaction (qPCR). d Samples were considered positive when the quantification cycle (Cq) value was ≤37 with a caveat that only samples with a Cq value ≤30 were selected for genetic typing. A multilocus variable number tandem repeat analysis (MLVA) typing assay was used to identify genetic variants. 2 The MLVA typing was based on the number of repeats of 2 M. hyopneumoniae variable number of tandem repeats (VNTRs): region repeat 1 (RR1) in the P97 locus and region repeat 3 (RR3) in the P146 locus. MLVA results were processed using commercial software, e and a minimum spanning tree was constructed to demonstrate the relationships among variants. To understand the interactions of M. hyopneumoniae within the swine production system, the distribution of M. hyopneumoniae variants, coughing scores, and the sow farms of origin were mapped to each room at each of the 3 farms (Fig. 1).
One hundred and 2 autopsies were performed during the study period. Ninety-two percent of the pigs that died presented lung lesions suggestive of pneumonia and 85% presented stomach ulcers at gross examination. Mycoplasma hyopneumoniae is not typically associated with mortality, but, in this farm, mortality was observed in combination with gastric ulcers and it is likely that gastric ulcers were the primary cause of death. Thus, the majority of the pigs with ulcers had pneumonia. The relationship between pneumonia and gastric ulcers is not well understood. It is not clear if gastric ulcers were exacerbated by pneumonia or vice versa. One theory is that pneumonic pigs have a poor overall health and reduced feed intakes and this potentially exacerbated the gastric ulcers. The gastric ulcers could have been caused by other factors, which were not investigated. An average of 5.6 samples was collected per room. One hundred bronchial swabs samples were positive for M. hyopneumoniae by qPCR (100/102; 98%). Twenty-eight tracheal aspirates were collected from rooms exhibiting dry cough but without mortalities and, from those, 24 were positive for M. hyopneumoniae by qPCR (24/28; 86%). Samples were not obtained from rooms where no coughing was observed and no mortalities were recorded (1 room on farm B and 2 rooms on farm C; Fig. 1). From the M. hyopneumoniae bronchial and tracheal qPCR-positive samples, 100 samples had a Cq value of ≤30 and therefore were selected for MLVA typing. Following MLVA typing, 95 variants were typeable, and 5 variants resulted untypeable. It is hypothesized that the untypeable variants may have been the result of samples containing a mixture of M. hyopneumoniae variants. While mixed MLVA types were confidently identified in a proportion of the samples, for the purpose of this investigation, the MLVA type detected with the highest intensity in the sample was selected as the predominant one and was used for analysis.
Four unique M. hyopneumoniae variants were identified in this production operation, namely MVLA variants 9-15, 11-21, 9-21, and 7-15. Overall, 1% of the typeable samples were variant 9-21 (n = 1), 27.4% were variant 11-21 (n = 26), 7.4% were variant 7-11 (n = 7), and 64.2% were variant 9-15 (n = 61). The number of repeats in P97 and P146 identified at these farms and the frequency of variant per room are shown in Figure 1. The MLVA variant 9-15 was the most prevalent, and was detected in 61 pigs and in 79% of rooms that tested positive for M. hyopneumoniae by qPCR. This variant was identified on all 3 farms, and it was the only variant found on farm C. MLVA variant 11-21 was detected in 26 pigs in 37% of the rooms on farms A and B. MLVA variant 7-15 was detected in 7 pigs in 2 rooms (11%) on farm A. Variant 7-15 accounted for all the samples tested in 1 room (room 4, farm A), as well as in room 9 of the same farm. A single sample of variant 9-21 was identified in 1 room on farm B, whereas all other samples from that room were variant 11-21. The minimum spanning tree constructed with all the variants detected in the 3 farms showed that variants appeared to be closely related to each other and to the reference strain (Fig. 2). Three variant clusters were observed: 1 cluster comprising 9 repeats in P97, another cluster comprising 21 repeats in P146, and a third cluster comprising 15 repeats in P146. Most samples across all farms belonged to a main variant cluster with 9 repeats in P97 (Fig. 2). These observations about relatedness of strains were made using a technique that uses only 2 loci, which represent a very small part of the genome that is being investigated; a higher resolution technique is needed to study the relatedness in more detail.
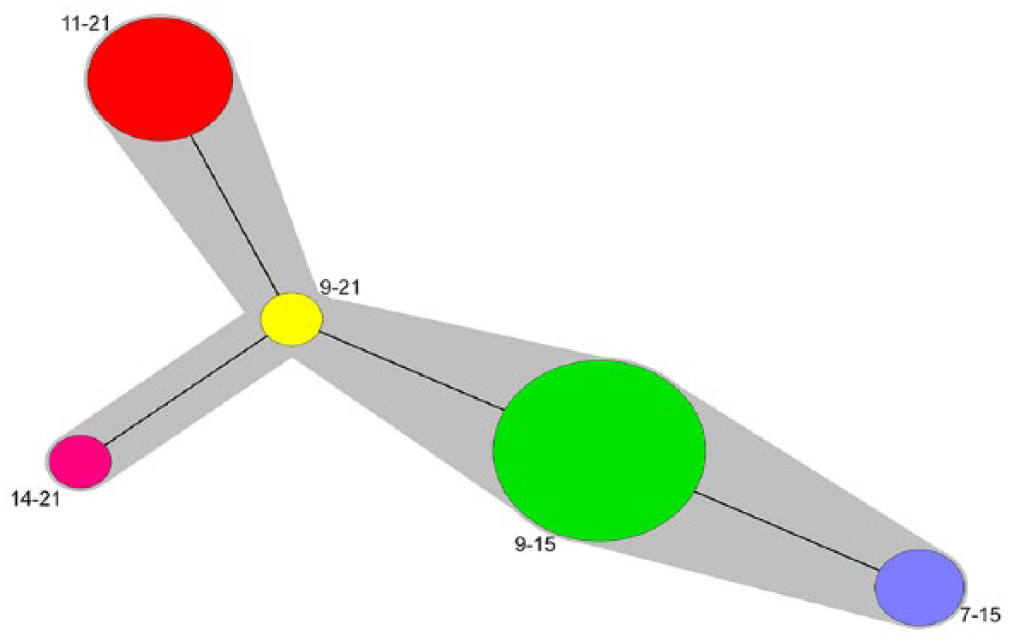
Minimum spanning tree showing 3 main Mycoplasma hyopneumoniae variant clusters. One variant cluster included clones with 9 repeats in the P97 loci, a second cluster included clones with 21 repeats in the P146 loci, and a third cluster included clones with 15 repeats in the P146 loci. Most samples across all farms belonged to a main cluster with clones with 9 repeats in the P97 loci. The relationships among farm variants in relationship to the U.S. 232 reference strain (type 14-21, and not found in the farms evaluated) are shown. The gray area surrounding the variants shows that the distance from one variant to another was not >1. Variant cluster connections are shown regardless of loci of common repeat.
The coughing assessment indicated that, out of the 22 rooms on the 3 farms, there were 9 rooms that had no coughing, 6 rooms that had some pens with ~2% of pigs coughing, and 7 rooms that had some pens with ~4% of pigs coughing (Fig. 1). Rooms that had pigs coughing had any of the 4 variants present. Rooms that did not have pigs coughing had either no M. hyopneumoniae detected or samples belonging to the 9-15 and 11-21 variants.
A graphic representation of the on-farm variant relationships, including clinical outcomes and room location is shown in Figure 1. On farm A, there were rooms with single M. hyopneumoniae variants from pigs originated from a single sow farm source (room 3) and from multiple sow farms (farm A, room 1). There were M. hyopneumoniae variants detected in rooms located within 15 m of each other that were completely different (farm A, rooms 3 and 4). There were rooms with M. hyopneumoniae variants found in pigs coughing located right next to rooms without coughing pigs (farm C, rooms 2 and 3). Only in 2 rooms (farm A, room 7 and farm B, room 3) did MLVA variant patterns suggest a combination of variants from adjacent rooms. The single sample of variant 9-21 detected on farm B may have been the result of an in situ recombination of variants 9-15 and 11-21. This recombination appears to have the 9 repeats at the P97 loci and the 21 repeats at P146 loci from both of the presumed parental variants. Because of the similarity in MLVA loci repeats, it can also be speculated that variant 7-15 is an on-farm mutation of variant 9-15. All 4 variants were found from bronchial swabs from dead pigs, and the 3 variants 9-15, 7-15, and 11-21 were found from tracheal samples. No obvious bacterial transmission pattern could be constructed tying variant to sow source, as sow farm samples were not available in this study.
The 4 variants identified in our study suggest limited M. hyopneumoniae genetic variation during the study period. The genetic variation detected in this investigation is similar to what has been reported in European farms. 11 This limited variation was found in spite of the fact that pigs had been comingled from multiple sow farms at weaning and had been together for 4 months. Nevertheless, enough genetic variation was found to be able to consider MLVA typing for epidemiological studies, to track how M. hyopneumoniae disseminates within and across farms, and to monitor genetic variability as a response of interventions like vaccination or treatment. A more extensive sampling protocol would be recommended in order to survey the entire herd including the sow farms of origin. Analysis of clinical signs, morbidity, and mortality would be necessary to determine if increased or decreased disease is associated with the observed variants. After the conclusion of the study, the veterinarian continued to rely on vaccination and implemented in-feed treatment protocols in finishing pigs to control M. hyopneumoniae clinical signs. The veterinarian plans to implement a M. hyopneumoniae elimination program. The cause of the gastric ulcers was not determined but the authors believe that alleviating M. hyopneumoniae clinical signs should improve feed intake and likely decrease gastric ulcer incidence.
Footnotes
Acknowledgements
We thank Dr. Douglas Marthaler for discussions on the molecular analysis and graphic representation of MLVA variant relationships.
Authors’ contributions
LG Pantoja contributed to conception and design of the study; contributed to acquisition, analysis, and interpretation of data; drafted the manuscript; critically revised the manuscript; and agreed to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved. K Pettit contributed to acquisition of data and drafted the manuscript. LF Dos Santos contributed to analysis and interpretation of data; drafted the manuscript; and critically revised the manuscript. R Tubbs contributed to acquisition of data. M Pieters contributed to design of the study; contributed to analysis and interpretation of data; drafted the manuscript; and critically revised the manuscript. All authors gave final approval.
a.
RespiSure One, Zoetis, Florham Park, NJ.
b.
BBL CultureSwab, BD Biosciences, Sparks, MD.
c.
Veterinary Diagnostic Laboratory, College of Veterinary Medicine, University of Minnesota, St. Paul, MN.
d.
VetMAX, M. hyopneumoniae; Applied Biosystems, Foster City, CA.
e.
Bionumerics version 7.2, Applied Maths Inc., Austin, TX.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The first author of this article (Lucina Galina Pantoja) is employed by Zoetis, the animal health company that sponsored this research.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was financially supported by Zoetis. Lucas Dos Santos was supported by Capes Foundation, Ministry of Education of Brazil (BEX17617/12-0).