
Abstract
Leptospires are excreted in the urine of infected animals, and the prompt detection of leptospiral DNA using polymerase chain reaction (PCR) is increasingly being used. However, contradictory data has emerged concerning the diagnostic accuracy of the most popular PCR assays that target either the 16S ribosomal RNA (rrs) or the subsurface lipoprotein (LipL32) genes. In order to clarify the effect of the gene target, a novel hydrolysis probe–based, quantitative real-time PCR (qPCR) assay targeting the LipL32 gene was developed, validated, and then compared directly to the previously described rrs hydrolysis probe–based qPCR using a convenience collection of canine urine samples. The novel LipL32 qPCR assay was linear from 5.9 × 106 to 59 genome equivalents per reaction. Both the LipL32 and the rrs qPCR assays showed a limit of detection of 10 target copies per reaction indicating an approximately equivalent analytical sensitivity. Both assays amplified all 20 pathogenic leptospiral strains tested but did not amplify a representative collection of bacteria commonly found in voided canine urine. When the field samples were assayed, 1 and 5 out of 184 samples yielded an amplification signal in the LipL32 and rrs assays, respectively. Nevertheless, when the limit of detection was considered as the cutoff for interpreting findings, the 4 discordant cases were judged as negative. In conclusion, our study confirmed that both LipL32 and rrs are suitable targets for qPCR for the detection of leptospiral DNA in canine urine. However, the rrs target requires the mandatory use of a cutoff value in order to correctly interpret spurious amplifications.
Introduction
Leptospirosis is a systemic zoonotic disease caused by pathogenic bacteria of the genus Leptospira. Both wild and domestic animals can be infected by leptospires and are reservoir hosts. During the acute phase, leptospires are present in different body fluids, such as urine, blood, cerebral spinal fluid, or the aqueous humor. 19 Animals recovering from leptospirosis may shed microorganisms in their urine for months or even indefinitely; therefore, active surveillance in animals is very important in order to prevent the spread of this zoonotic disease. 3 The detection of leptospiral DNA using a polymerase chain reaction (PCR) assay is increasingly recommended as a suitable ancillary assay for the early detection and diagnosis of chronic carriers in companion animals.16,17,36
Quantitative real-time PCR (qPCR) is increasingly being used over traditional PCR owing to its robustness and low turnaround time. In the past decade, 2 qPCR assays using double-stranded DNA–binding dye, one targeting a gene that codes for a subsurface lipoprotein (LipL32) and the other targeting the LA0322 locus, have been validated in humans.20,21 Double-stranded DNA–binding dyes are popular because of their lower cost as compared with hydrolysis probe technologies but, on the other hand, they can be intrinsically less specific. 9 The inclusion of fluorescently labeled probes is warranted in real-time PCR assay design. 37
A hydrolysis probe–based qPCR assay targeting the 16S ribosomal RNA (rrs) gene has also been validated in humans.28,30 This assay has gained popularity over time and has also been adopted for use in dogs 22 as the assay has been validated for all pathogenic leptospires, including those known to infect dogs.
In the past 5 years, the accuracy of the rrs qPCR assay has been questioned because of specificity concerns,13,31,35 despite the fact that a thorough in vitro validation was carried out in the original study. 30 Nevertheless, in a case-control study, the rrs qPCR assay was shown to have a greater sensitivity and a similar specificity, in direct comparison with the LipL32 qPCR. 33 However, the issue is still under debate as to the identity of the cross-reacting bacteria and whether the rrs specificity is adequate.
The same concerns are even more compelling in dogs. Despite the satisfactory data regarding in vitro accuracy, data regarding clinical cases and the use of qPCR, using both rrs and LipL32 targets in natural canine infections, is scarce. 22 This is also true when interpreting the findings in those studies that used different qPCR assays when investigating the molecular epidemiology of leptospirosis in dogs. 27 Ideally, the use of the rrs gene, which is largely used for classification purposes,10,23 has the advantage of a wider database of nucleotide sequences for in silico validation. On the contrary, because it is a well-conserved gene among many bacterial species other than leptospires, the rrs gene is more prone to unexpected cross-reactivity primarily when the assay is applied to urine and similar specimens that contain poorly characterized polymicrobial flora.13,35 However, LipL32 is a subsurface protein that, to date, has been found exclusively in pathogenic leptospires.4,14,24,26 Hence, while the rrs qPCR amplifies pathogenic, and also intermediate pathogenic, leptospires, the LipL32 qPCR detects only the pathogenic leptospires.14,31,33
In order to clarify the effect of the gene target in detecting leptospires in canine urine, in the present study, a novel hydrolysis probe–based qPCR assay targeting the LipL32 gene was developed and validated in vitro. The novel assay was then directly compared with the rrs gene hydrolysis probe–based qPCR assay (rrs qPCR) in a convenience collection of canine urine samples that represented the most common matrix investigated for the assessment of the potential hazard of leptospire shedding in a veterinary clinical context.
Materials and methods
Sample collection
A total of 184 urine samples were obtained from dogs undergoing routine urinalysis at the Veterinary Clinical Pathology Service of the University of Bologna (Ozzano dell’Emilia, Bologna, Italy) between September and December 2012. The urine samples were included in the study regardless of the method of collection (free-catch, cystocentesis, or catheterization; Table 1). The urine samples were collected in sterile containers and refrigerated at 5°C ± 3°C for a maximum of 48 hr before DNA extraction.
Results of the quantitative real-time polymerase chain reaction (qPCR) assay based on the 16S ribosomal RNA gene (rrs) and relationship with the collection method.*
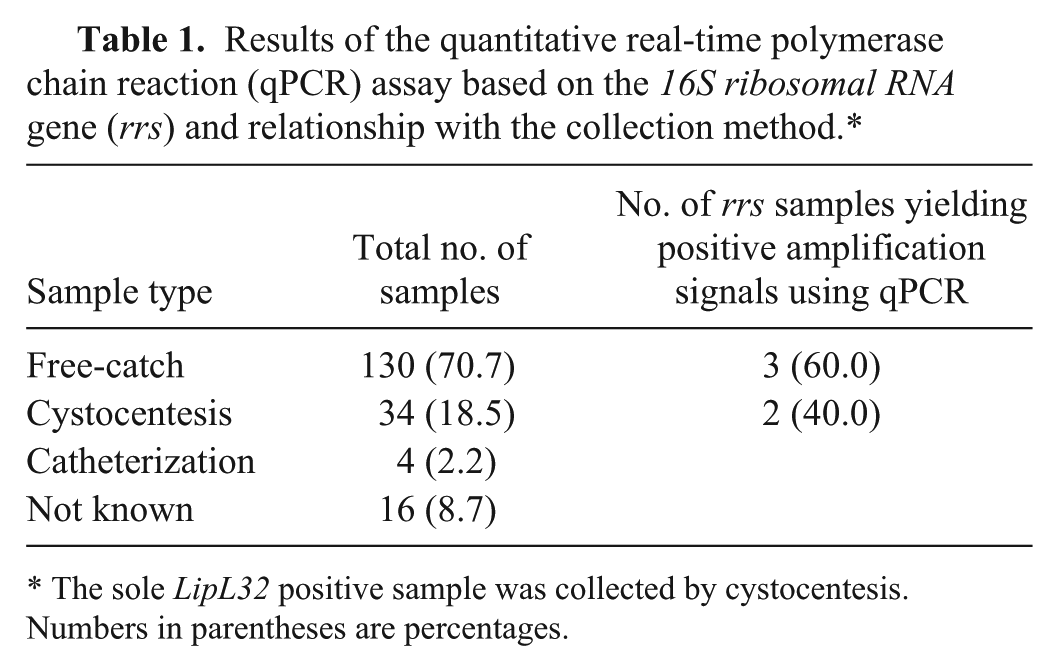
The sole LipL32 positive sample was collected by cystocentesis. Numbers in parentheses are percentages.
DNA extraction method from urine samples
Genomic DNA (gDNA) was extracted from the urine specimens as described previously, 16 with minor modifications. Briefly, 1.5 mL of the urine sample was centrifuged using a standard fixed rotor benchtop centrifuge a (800 × g, 10 min) at room temperature; the supernatant was then transferred to a new tube and centrifuged again (12,100 × g, 30 min). The resultant pellet was resuspended in 200 µL of phosphate buffered saline. The DNA was immediately extracted from the pellet using a silica-based column method of purification, b according to the manufacturer’s instructions, and stored at −20°C.
Evaluation of PCR inhibitors
To check for the presence of PCR inhibitors left after the nucleic acid purification procedure, an exogenous internal control (EIC) was added directly to the sample, and the presence of the EIC was confirmed by PCR. This approach was preferred rather than the use of an endogenous internal control because the double centrifugation method used for processing urine markedly reduces the content of somatic cells and of endogenous gDNA in the samples tested. Hence, in our study, a mimic EIC composed of an artificial sequence used in another study was utilized. 34
Bacterial strains
To accomplish the majority of the validation experiments, Leptospira serovar Pomona strain Mezzano I and serovar Canicola strain Alarik, provided by the Animal Leptospirosis National Reference Center of the Lombardy and Emilia Romagna Experimental Zootechnic Institute (IZSLER; Brescia, Italy), were used. To assess the ability of the PCR assays to detect other serovars, 18 leptospiral strains provided by a clinical laboratory and belonging to different serovars were used. All the strains are listed in Table 2. In addition to leptospires, 5 other reference bacterial strains, shown in Table 3, were used to evaluate assay specificity. The DNA from 200 µL of leptospiral culture was extracted using a silica-based column method b as recommended by the manufacturer.
Leptospiral strains used in the current study.

Bacterial strains and their respective DNA concentrations in the samples used to test the specificity of the hydrolysis probe–based real-time polymerase chain reaction assays.

ATCC = American Type Culture Collection.
Reference standard
Leptospiral gDNA samples purified from the pure cultures were quantified using a fluorescence-based nucleic acid quantification method. c The gDNA of Leptospira interrogans serovars Canicola strain Alarik and Pomona strain Mezzano I was subsequently serially diluted 1:10 in a solution of molecular biology grade–water containing 10 ng/µL of canine gDNA and the EIC at equimolar concentration with respect to the urine samples.
Furthermore, both the almost complete 16s rRNA gene and a partial sequence of the LipL32 gene were amplified, gel purified, and cloned into a commercial vector. d The presence of the inserts was verified using M13 forward and reverse primers, and sequenced using a commercial kit e and an automated sequencer. f The primers used for amplifying the rrs and LipL32 targets for cloning were as follows: 27F_Universal (5′-AGAGTTTGATCMTGGCTCAGF-3′), 1492R_Universal (5′-GGTTACCTTGTTACGACTT-3′), fwdclon_LipL32 (5′-TGCAAGCATTACCGCTTGTGGTGC-3′), and Revclon_LipL32 (5′-TGAGTGGATCAGCGGGCTCACAC-3′), respectively. The plasmid copy number was determined by spectrophotometry. The plasmids were then linearized with NcoI, g and were serially diluted in the same manner as the gDNA.
The validation approach was carried out according to the methods implemented for qPCR. The analytical sensitivity used as the lower limit of detection (LOD) was experimentally estimated as the last serial dilution investigated yielding positives on all 5 replicates. This translates into a probability of testing positive >50% with a 95% level of confidence.6,7 The highest quantification cycle (Cq) value observed in the serial dilution corresponding with the LOD was then considered as the cutoff value for interpreting the findings as positive or negative. 7 The Cq was standardized between different runs by manually setting a common quantification value. The quantification fluorescence level was set at a value that crossed the sigmoid curve in the log-linear exponential phase in all different runs, as was visually ascertained.
qPCR assay design
The primers and probes were designed with software h using a consensus sequence from the alignments of available Leptospira spp. LipL32 gene partial sequences were obtained from the GenBank nucleotide sequence database. The primers used were as follows: Fwd_LipL32 (5′-TCCCAGGGACAAACGAAACCGT-3′), Rev_LIPL32 (5′-TGTTTCCATCGGCTAAACCGT-3′), and LipL32_probe (5′-[6FAM]ACGTAAAGCCAGGACAAGCGCCG[BHQ1]-3′). The oligonucleotides were purchased from a commercial source. i The primers and probes of the rrs assay were those reported previously with the difference that the quencher dye was represented by black hole quencher 1 and not by the tetramethylrhodamine fluorophore as originally reported. 30
PCR conditions
The LipL32 qPCR was carried out using a mixture of 10 μL of 2× PCR mix, j 900 nM each of the forward and reverse primers, 400 nM of probe, 2.5 μL of template, and molecular biology–grade water to reach a final volume of 20 μL. The qPCR was carried out using a 4-step protocol: initial denaturation at 95°C for 4 min followed by 50 cycles at 94°C for 30 sec, 57°C for 15 sec, 57°C for an additional 15 sec with signal acquisition and at 72°C for 25 sec in a thermal cycler. k The rrs qPCR assay contained the same final concentrations of reagents with the sole difference that the probe was 300 nM. The cycling conditions were identical to those reported previously. 30 Positive and negative controls were included in each qPCR run.
PCR assays for possible interfering bacteria
To test the hypothesis that other bacteria lining the urinary tracts of dogs as normal colonizing microflora (e.g., Lactobacillus spp. and Atopobium spp.). 13 or those causing urinary tract infections (e.g., Enterococcus spp.)15,25 could be responsible for spurious amplifications by an rrs qPCR assay, 13 a subset of urine samples was tested using a qPCR with a double-stranded DNA–binding dye l and genus-specific primers for the presence of Lactobacillus spp., 38 Enterococcus spp., 18 and Atopobium spp.8,13 The subset of samples was composed of the 5 samples that originally gave amplification signals in the rrs qPCR assay as well as 40 samples chosen at random after stratification according to the method of collection.
Results
Using the serial dilutions of gDNA from leptospires of the serovar Canicola, the LipL32 qPCR assay showed a broad dynamic range achieving linearity from 5.9 × 106 to 59 genome equivalents (GE) per reaction. The R2 coefficient was 0.998, and the PCR efficiency was 103.94%. With gDNA from leptospires of the serovar Pomona, the R2 coefficient of linearity was 0.998 between 6.1 × 106 and 61 GE per reaction, and the PCR efficiency was 100.58% (Fig. 1).
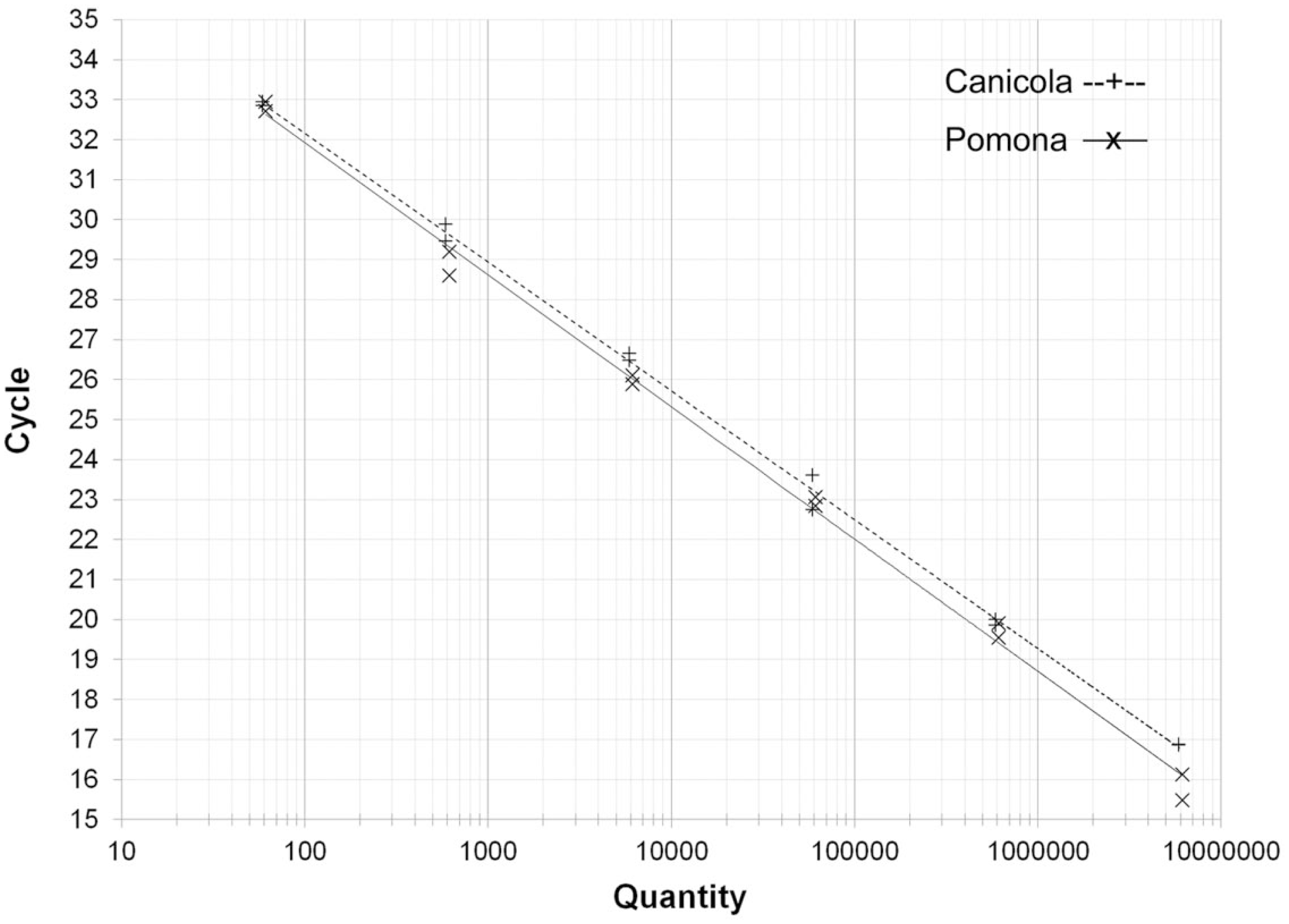
Linearity and calibration curve of the LipL32 quantitative real-time polymerase chain reaction assay using genomic DNA of Leptospira interrogans serovars Canicola and Pomona as templates.
Five replicates of 2 standards containing 4.0 and 0.4 copies/µL, respectively, were assayed using the LipL32 and the rrs assays to establish the LODs. Specifically, 10 copies per reaction were detected in all replicates, while the 1.0 copy reactions were found to be positive in 3 replicates and 1 replicate in the LipL32 and the rrs assays, respectively.
The quantification values were manually set at 0.02 and 0.05 log in the LipL32 and the rrs assays, respectively. Using the above-mentioned settings, the Cq of the LOD for LipL32 varied from 33.1 to 35.4 (mean 34.8); thus, the cutoff value was set at 36. The Cq of the LOD for the rrs assay varied from 35.0 to 36.4 (mean 35.8); accordingly, the cutoff value was set at 37. All 18 leptospiral strains tested at a concentration of ~1 to 8 × 102 GE gave positive signals with both qPCR assays. When challenged against the bacterial strains in Table 3 with GE ranging from 1.4 × 107 to 5.4 × 107, no signals were produced in 2 replicates with either qPCR assay.
The EIC added to the field samples was detected in all cases, confirming the adequacy of the nucleic acid purification technique and the absence of inhibitors in the PCR runs. The linearity and LOD experiments had already demonstrated that the EIC did not interfere with the assays. The field samples gave the following results: 1 out of 184 and 5 out of 184 yielded positive signals with the LipL32 and the rrs assays, respectively. One case was found to be positive concordantly with both PCR assays. Samples yielding amplification signals were retested in duplicate in a different experiment run, again yielding a signal in at least 1 of the replicates. Nevertheless, when the cutoff values were considered, 4 samples were found to have spurious amplification with a Cq higher than that of the cutoff (Fig. 2).
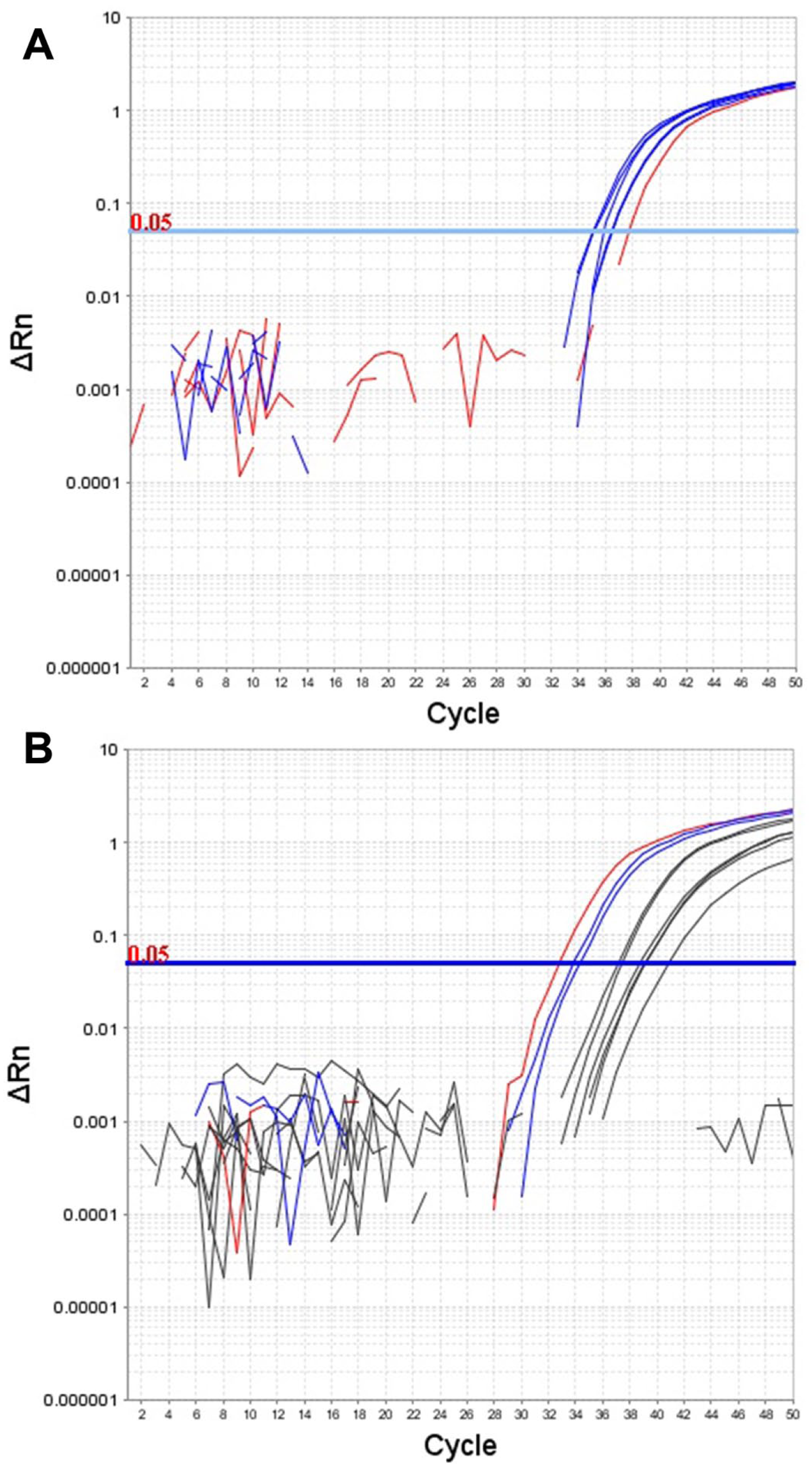
Analytical sensitivity experiments for the 16S ribosomal RNA (rrs) quantitative real-time polymerase chain reaction assay.
The rrs PCR spurious positive amplifications were not significantly associated with the sampling method (Fisher exact test; P = 0.28; Table 1). As a result of the concordant findings of both assays, the prevalence observed was estimated at 0.5% (confidence interval: 0.1–3.0%).
Forty-five samples, including the 5 samples that yielded amplification signals in at least 1 leptospiral PCR assay and an additional 40 samples, assayed with genus-specific PCR for Lactobacillus spp., Enterococcus spp., and Atopobium spp., showed 2 positive findings for Enterococcus spp. and 1 positive finding for Atopobium spp. None of the samples that yielded amplification signals in either the rrs or the LipL32 qPCR assays was positive for interfering bacteria in any of the PCR assays.
Discussion
In addition to the obvious diagnostic relevance, the detection of the urinary excretion of leptospires embodies a zoonotic risk for owners and veterinary practitioners. Studies have questioned the accuracy of the hybridization probe–based qPCR assay for the detection of pathogenic leptospires.31,35 Indeed, all of the hybridization probe–based qPCR assays described to date were thoroughly evaluated under laboratory conditions but not all of them had undergone “in field” validation. Because a true reference method for the detection of pathogenic leptospires in dog urine does not exist, the present study was aimed at developing a novel LipL32 qPCR assay using a hybridization probe, and comparing the performance of that assay with the qPCR assay targeting the rrs. 30
As regards analytical sensitivity, both assays performed well, reaching a consistent LOD of 10 target copies per reaction. Furthermore, all of the leptospiral strains tested were readily detected by both assays at levels as low as 100 GE per reaction. It is known that slightly different LOD values can result when the same assay is run using different thermal cyclers. 31
The analytical specificity was also adequate under laboratory conditions. Indeed, neither assay gave a signal when representative bacteria commonly colonizing the canine urinary tract were tested as templates at levels of >1 × 107 GE. Other bacteria found to interfere with leptospiral PCR assays when used on human urine, such as Peptostreptococcus stomatis and Peptostreptococcus anaerobius, were not available and were not examined in our study owing to the fact that they were not examined in other veterinary studies.
When the assays were used to test 184 canine urine samples, the rrs qPCR assay yielded spurious amplification with Cq values above the cutoff value in 4 cases. Attempts to sequence these samples were unsuccessful, and only faint smears were observed when PCR products were run in an agarose gel. In an attempt to investigate the possibility of cross-reactions with other microorganisms found in the urinary tracts of dogs, all of the urinary samples were screened for the presence of bacteria normally colonizing the urinary tract (e.g., Lactobacillus spp. and Atopobium spp.) or those possibly causing inflammation of the urinary tract (e.g., Enterococcus spp.). A very low prevalence of these bacteria was found, and no samples giving amplification signals in the leptospiral rrs qPCR were found to be positive for these bacteria. It is possible that the method of DNA purification, which included a centrifugation step, could explain this situation. The centrifugation step could reduce the number of large bacteria, such as lactobacilli, which could be pelleted with the somatic cells and then discarded. However, using a different method, without a preliminary centrifugation step, could add interfering bacteria to the pellet used for the DNA purification. One sample was positive according to both assays and showed a Cq close to the LOD, emphasizing the importance of a very sensitive assay to screen canine urine for the presence of leptospires. Similar findings of false-positive results in the rrs qPCR as a result of bacteria of commensal and/or environmental origin have been reported in a 2015 study. 11
The likelihood of false-negative results in the LipL32 assay was very low based on the ability of the assay to detect the entire collection of leptospiral strains tested and the LODs observed. However, intermediate pathogenic strains were not among the strains tested in the current study and they would not have been detected using the LipL32 targeting assay as these species lack the LipL32 gene.31,33 The pathogenicity of these intermediate strains, which form a separate clade between the pathogenic and saprophytic leptospires,23,31,33 is controversial. The only species with intermediate pathogenicity reported in dogs is Leptospira wolffii in Iran and India.29,39 Evidence of a level of pathogenicity of L. wolffii for humans has been shown.2,39 Therefore, also in western countries, the possibility of the presence of L. wolffii in canine urine warrants additional study.
The assessment of the prevalence of urinary excretion of leptospires was not an aim of our study. However, it is necessary to comment on the observed prevalence because it is notably lower than the prevalence data (6.83–22.0%) reported in previous studies regarding dogs.17,27,39 In fact, the findings of the current study should not be considered a reliable indicator of the actual prevalence of leptospirosis for 3 reasons. First, a collection of convenience samples is not a truly random set of samples and does not reflect the general Italian dog population. However, convenience samples usually represent the animal population that is seen by veterinarians and could represent the best way of measuring the relative risk in a particular clinical setting. 32 Second, the Veterinary Teaching Hospital of the University of Bologna primarily admits animals that have already been treated, in particular, with antibiotics, by referring practitioners. Thus, it is likely that leptospirosis-affected dogs that were not excreting leptospires were also included in our study as previously reported. 12 Finally, leptospirosis in a low endemic area shows a seasonality that is also influenced by rainfall. 19 The likelihood that a cluster of cases of leptospirosis was not included in the limited period of time during which the collection of samples was carried out in this study is relevant.
Conversely, the possibility of PCR inhibition could be ruled out as the cause of the lower prevalence observed with respect to previous studies. In fact, urine represents a very useful specimen for the detection of leptospires because the bacterial DNA can be found in the urine of dogs much longer than in blood,1,5,35 provided that the risk of inadequacy of the sample due to the presence of the PCR inhibitors present in urine is adequately taken into consideration and managed. The use of spiked nonrelated DNA (EIC) adds validity and strengthens the consistency of the findings.
In conclusion, our study found that both rrs and LipL32 were suitable targets for hybridization probe–based qPCR assays, although the former required comprehensive validation because weak amplifications could occur. These weak amplifications will show up as spurious results unless a suitable cutoff value is established during the validation of the assay.
Footnotes
Acknowledgements
The inactivated leptospires of serovars Leptospira Pomona strain Mezzano I and Leptospira Canicola strain Alarik were donated by Dr. Silvia Tagliabue. The other inactivated leptospiral strains were donated by Dr. Francesca Brunello.
Authors’ contributions
F Gentilini and ME Turba contributed to conception and design of the study. F Gentilini contributed to acquisition, analysis, and interpretation of data, and critically revised the manuscript. E Zambon contributed to analysis of data. RG Zanoni contributed to analysis and interpretation of data, and critically revised the manuscript. ME Turba contributed to interpretation of data. All authors drafted the manuscript; gave final approval; and agree to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
Mini Spin, Eppendorf Srl, Milan, Italy.
b.
NucleoSpin tissue, Macherey-Nagel GmbH & Co. KG, Düren, Germany.
c.
QuantiFluor dsDNA system, Promega Italia Srl, Milan, Italy.
d.
Life Technologies PCR 2.1-TOPO vector, Thermo Fisher Scientific, Monza, Italy
e.
Life Technologies Big Dye Terminator v1.1, Thermo Fisher Scientific, Monza, Italy.
f.
ABI Prism 310, Thermo Fisher Scientific, Monza, Italy.
g.
EuroClone SpA, Milan, Italy.
h.
Life Technologies Primer Express, Thermo Fisher Scientific Inc., Waltham, MA.
i.
Proligo, Sigma-Aldrich Srl, Milan, Italy.
j.
Fermentas Maxima probe master mix, Thermo Fisher Scientific, Monza, Italy.
k.
Life Technologies StepOne, Thermo Fisher Scientific, Monza, Italy.
l.
Fermentas Maxima SYBR Green qPCR Master Mix, Thermo Fisher Scientific, Monza, Italy.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by an RFO 2011 grant from the University of Bologna.