
Abstract
In the present study, a TaqMan quantitative polymerase chain reaction (qPCR) assay for detecting the 16S ribosomal RNA gene of Lawsonia intracellularis in porcine native ileal mucosal scrapings, fecal samples, and formalin-fixed and paraffin-embedded (FFPE) ileal samples is described. Samples from 62 pigs were examined. The results of the qPCR were compared with results obtained with conventional detection methods (PCR, immunohistochemistry, in situ hybridization, and silver staining) from a previous study and correlated well. The qPCR assay proved to be very sensitive and specific. In particular, the sensitivity of TaqMan PCR was significantly higher than conventional PCR on FFPE tissues because of a much shorter amplicon. A higher number of copies per gram of sample material was detected in native mucosa and FFPE tissue compared with feces, especially in highly positive animals. The detection limit for the qPCR was at 4 copies per well in native mucosal scrapings and 18 copies per well in feces and FFPE tissue, respectively. Inhibition of the qPCR reaction was checked by simultaneous detection of a recombinant beta-actin plasmid using a second fluorescent probe. A decreased signal from this internal control plasmid revealed inhibition of the PCR reaction in 21% of native mucosal samples and 1.6% of fecal samples. With a 10-fold dilution of template, the inhibition could be overcome.
Keywords
Lawsonia intracellularis is the etiologic agent that causes porcine proliferative enteropathy, a transmissible enteric disease with a high prevalence in growing pigs world-wide. 6,10 Since L. intracellularis is an intracellular bacterium, diagnosis by bacterial culture is not possible. Necropsy and histopathological techniques including immunohistochemistry (IHC) and in situ hybridization (ISH) are useful for the identification of active disease but are less sensitive than polymerase chain reaction (PCR) in subclinically infected animals. 4 Several PCR and quantitative PCR (qPCR) assays with different sensitivities have been established for the detection of L. intracellularis mainly for fecal samples, including an extensively validated, recently published assay. 3,7,9 Although a higher number of L. intracellularis would be expected in mucosal tissue than in feces, a comparative study showed a higher sensitivity of PCR in feces than in tissue samples. 4 In that study, a well-established PCR assay 3 produced amplicons with a length of 319 bp. In formalin-fixed and paraffin-embedded (FFPE) tissue, the DNA generally shows substantial fragmentation, leading to a decreased sensitivity of PCR assays with relatively long amplicons. 1,11 The objectives of the current study were 1) to establish a TaqMan qPCR assay for detection of L. intracellularis that is similarly sensitive in native ileal mucosal scrapings, fecal samples, and FFPE ileal samples; 2) to quantify the amount of L. intracellularis in the samples; 3) to check the samples for inhibition simultaneously; and 4) to compare the results with those previously obtained with conventional detection methods.
Ileal and fecal samples had been taken at necropsy from 204 pigs aged 1–6 months with a history of diarrhea and/or retarded growth in the course of a previous study. 4 All samples had been tested for the presence of L. intracellularis using conventional PCR, IHC, ISH, and silver staining, and 52 pigs had tested positive with 1 or more of these techniques. DNA extracts from native ileal tissue from these 52 animals plus 10 animals that had tested negative were used for the current study. From 18 of these 62 pigs, DNA extracts from feces and FFPE ileal tissue were also used. All DNA extracts were produced with commercial extraction kits. a,b , 4
A primer-probe combination specific for the 16S ribosomal RNA (rRNA) gene of L. intracellularis was chosen using Primer Express software version 2.0. c The sequences were forward primer 5′-GTTCCCGGGCCTTGTACAC-3′, reverse primer 5′-CCGGCTTTGGGTAAAACCA-3′, d and TaqMan probe 5′-FAM-CCGCCCGTCACACCACGAAA-TAMRA-3′, e resulting in an amplicon of 61 bp. As determined by BLAST search (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi), this primer-probe combination was 100% identical to 5 sequences of the 16S rRNA gene of Lawsonia/Ileal symbiont intracellularis from pigs (GenBank accession nos. EU348664, AM180252, U65995, U30147, L15739) as well as to 1 Ileal symbiont intracellularis from a golden hamster (Mesocricetus auratus; accession no. U06423) and to 1 intracellular bacterium from a ferret (Mustela putorius furo), the sequence of which is 100% identical to L. intracellularis (accession no. U30147), although it has been designated as Desulfovibrio sp. (accession no. U07570). Primers were tested on a Lawsonia-positive fecal sample for formation of polymers with an SYBR Green assay, f resulting in a single melting point at dissociation.
To generate a standard curve, the above-described amplicons were achieved from a PCR-positive fecal sample. These were cloned into competent Escherichia coli using a commercial system g and checked by nucleotide sequencing. h,i Concentration of extracted DNA j was determined photometrically, and the number of plasmids was calculated. Serial dilutions were used to generate a standard curve to optimize primer and probe concentrations. A standard curve was included in each subsequent run and showed a repeatability between 89.6% and 93.3% between runs. The detection limit in serial plasmid dilutions was found to be 6 plasmids per well.
Plasmids containing a recombinant insert with a total length of 101 bp consisting of the above primer sequences at the ends and a centrally located 63-bp amplicon from equine beta-actin were constructed, checked, and diluted as described above. They were used as internal control (IC) analogous to previous description. 2 A TaqMan probe was designed complementary to a part of the beta-actin insert (5′-JOE-TTCACCACCACGGCCGAGAGG-TAMRA-3′). k
In the optimized protocol, each qPCR well contained 25 μl of reaction mix composed of 12.5 μl of Master Mix, 1 600 nM (1.5 μl) of forward and reverse primers each, 250 nM (2.5 μl) Lawsonia and beta-actin probes each, 100 (2 μl) IC plasmids, and 2.5 μl of template, which consisted of DNA extracts (template or standards) or laboratory-grade water in no template controls (NTC), respectively. In the case of suspected inhibition of the qPCR reaction, the reaction was repeated with a 10-fold dilution of the DNA extract as template. The thermocycling conditions consisted of an initial step of 50°C for 2 min and 95°C for 10 min, followed by 45 cycles of 95°C for 15 sec and 60°C for 1 min. All NTC remained negative throughout the whole study. Standards, NTC, and samples were all tested in duplicate.
The amplification, signal detection, and data analysis were conducted with a commercial real-time PCR system c using automatic threshold settings. The cutoff value for detection of Lawsonia genes was set at a threshold cycle (Ct) of 40. Results were compared with those achieved with the above-mentioned conventional techniques from the previous study. For descriptive statistical analyses, a commercial spreadsheet program was used. The efficiency of the complete assay including 100 IC plasmids per well was 99.68%, as calculated from the standard curve that was generated from 10-fold diluted solutions of Lawsonia plasmids. The R 2 value was 0.984.
An amount of 100 IC plasmids gave a constant and positive signal without inhibiting the reaction efficiency of the Lawsonia assay. In cases of very high Lawsonia numbers in the sample, the signal of the IC was reduced or absent. Inhibition indicated by slight reduction to total absence of IC signal was seen in 13 mucosal samples (21%) and 1 fecal sample (1.6%). In these samples, a 10-fold dilution of the template resulted in a more than 10-fold increased copy number per gram sample or the appearance of a signal in an originally negative sample.
Results obtained with qPCR from different sample types from 1 animal, respectively, correlated well with each other. The calculated copy number in DNA extract of 1 g native mucosa compared with the copy number in the DNA extract of 1 g FFPE tissue varied within 1 order of magnitude. However, because of the small amount of initial sample mass in FFPE tissue (about one third of native mucosal mass), the assay was less sensitive in this material. The calculated copy number in DNA extract of 1 g feces was approximately 2 orders of magnitude less than in mucosal samples in highly positive animals and approximately 1 order of magnitude less in slightly positive samples (Fig. 1). This can be explained with the intracellular location of L. intracellularis and a subsequently higher number in tissues than in feces.
The lowest detected copy number per well was 4 copies twice in mucosal scraping with a Ct of 38.13 and 39.79 corresponding to 32 × 103 copies in DNA extract of 1 g sample and 18 copies in feces and FFPE material with a Ct of 37.22 and 37.25 corresponding to 7.2 × 103 and 432 × 103 copies in DNA extract of 1 g sample, respectively. Since the complete genome of L. intracellularis as available via GenBank (accession no. NC_008011) contains 2 copies of the 16S rRNA gene, the actual number of genome equivalents or bacteria can be calculated by dividing the copy number by 2. However, it must be kept in mind that although extraction efficiency was not tested in the current study, previous studies have determined a recovery of 3.5% for genomic DNA from feces using the same commercial kit as in the present study. 8
Results of qPCR from all 62 examined animals correlated well with results previously obtained with conventional diagnostic methods. 4 Of the 18 animals examined with qPCR in different sample types, the 2 animals with the highest copy number had also tested positive with all conventional methods. With decreasing copy number, results became negative with these methods in the following order: silver staining, ISH, IHC, PCR from FFPE material, PCR from native mucosa, and PCR from feces (Table 1). The copy number in the 44 animals examined with qPCR only in native mucosa showed a similar correlation with results from conventional methods as the above 18 animals in native mucosa. In all 62 animals, native mucosal samples that had tested positive with conventional PCR showed a tendency toward low Ct values in qPCR, while those tested negative gave mostly high Ct values. Two animals were positive with both qPCR and conventional PCR only in fecal samples. Since both assays target different gene loci, an unspecific signal obtained with both methods is highly unlikely. One possible explanation is that ileal samples in these cases did not contain the infected area of intestine, as L. intracellularis can lead to multiple small foci of infection or may also infect colonic mucosa. 5 Especially in FFPE ileal samples, the examined tissue may not well represent the whole length of ileum. Similar cases were seen in a study comparing qPCR of fecal samples with IHC on ileal sections. 7 Another explanation might be the passage of bacteria through the intestine without any infection, although this has never been reported in experimental studies.
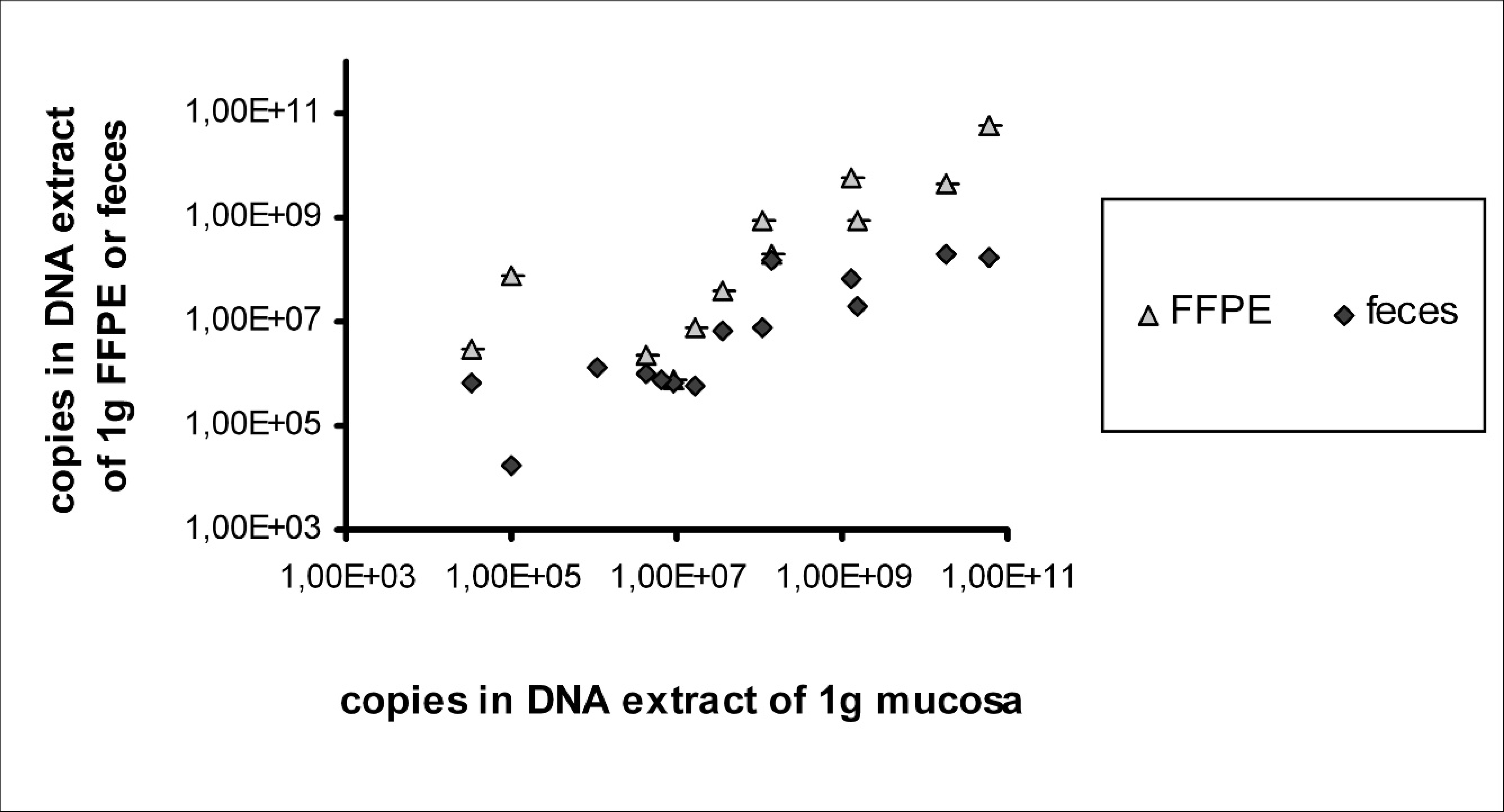
Positive results from quantitative polymerase chain reaction: correlation of copy number per gram native mucosa with copy number per gram feces and formalin-fixed, paraffin-embedded (FFPE) ileum, respectively.
After comparative analysis, the detection limits of conventional PCR were calculated as 64 copies per tube or 2 μl of template for mucosa, 34 copies per tube for feces, and 28,800 copies per tube for FFPE material. The high detection limit of the conventional PCR assay in FFPE tissue compared with native mucosal and fecal samples is very likely due to DNA fragmentation during the fixation and embedding process. As previously demonstrated, 11 PCR assays with amplicons of more than 500 bp consistently yield negative results when applied to FFPE tissue samples that test positive with PCR assays with shorter amplicons. Thus, the amplicon of 319 bp from conventional PCR in this case 4 might lead to decreased sensitivity, although it is still detectable in highly positive samples. This explanation is supported by the results of the qPCR assay, with an amplicon length of 61 bp being much more sensitive in FFPE material with a detection limit equal to that of fecal samples.
Differences in sensitivity of diagnostic methods and sample material. *
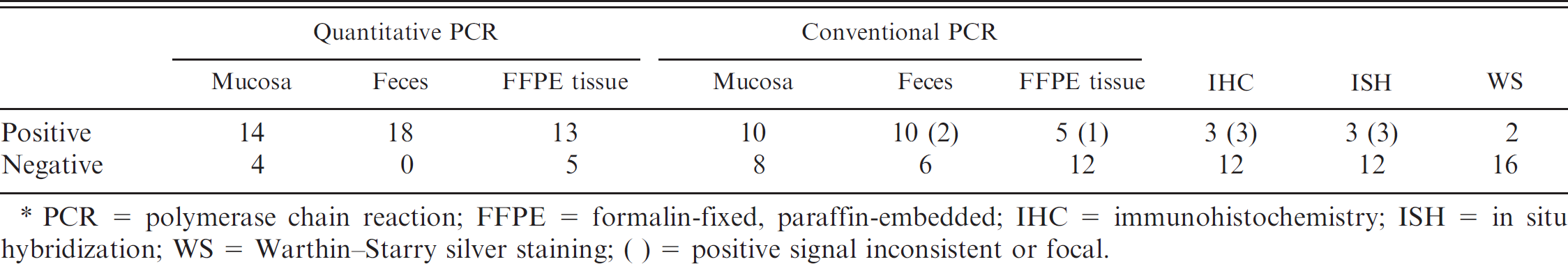
PCR = polymerase chain reaction; FFPE = formalin-fixed, paraffin-embedded; IHC = immunohistochemistry; ISH = in situ hybridization; WS = Warthin–Starry silver staining; ( ) = positive signal inconsistent or focal.
From the 13 samples showing inhibition of the qPCR from mucosa, 5 samples were positive with conventional PCR. On the other hand, of 7 samples that had tested negative with all conventional methods, 5 samples gave positive signals in qPCR from mucosa (inhibition in 2 cases), 3 in qPCR from feces, and 2 in qPCR from FFPE material (both without inhibition). The simultaneous detection of 100 recombinant plasmids as IC proved to be a useful tool, revealing a significant level of inhibition especially in mucosal samples, although these had been cleansed carefully during sampling. 4 The above results suggest a greater susceptibility to inhibition in qPCR from native mucosa in contrast to a decreased susceptibility to inhibition in qPCR from feces compared with conventional PCR. An influence might be attributed to the different extraction methods used for mucosal samples and feces in this case.
In the current study, a very sensitive and specific TaqMan qPCR assay for the detection of L. intracellularis in native ileal mucosal scrapings, fecal samples, and FFPE ileal samples from pigs is described. The results correlated well with a previous examination of the same samples using conventional PCR, IHC, ISH, and silver staining. The sensitivity could be increased compared with conventional PCR, especially in FFPE tissue. The simultaneous detection of an IC plasmid revealed significant inhibition in 13 mucosal samples and 1 fecal sample, which could be overcome with 10-fold dilutions of the template. This assay may also be very useful for clinical studies as the quantity of organisms is correlated with clinical signs and pathological lesions. Thus, the quantity of bacteria in the feces indicates the clinical relevance of the infection.
Acknowledgements. The invaluable work of Anton Maderner and Klaus Bittermann is gratefully acknowledged. The authors also thank the staff members of Clinical Virology, University of Veterinary Medicine Vienna, for their technical support.
Footnotes
a.
Genomic DNA Isolation Kit Tissue & Cells, Nexttec Biotechnologie, Leverkusen, Germany.
b.
QIAamp® DNA Stool Mini Kit, Qiagen, Hilden, Germany.
c.
Applied Biosystems 7300 Real-Time PCR System, Applied Biosystems, Foster City, CA.
d.
Invitrogen Corp., Carlsbad, CA.
e.
Microsynth AG, Balgach, Switzerland.
f.
SYBR Green PCR Master Mix, Applied Biosystems, Foster City, CA.
g.
pGEM®-T Easy Vector System, Promega Corp., Madison, WI.
h.
ABI Prism Big Dye Terminator cycle sequencing ready reaction kit, Applied Biosystems, Foster City, CA.
i.
ABI Prism 310 genetic analyzer, Applied Biosystems, Foster City, CA.
j.
Pure Link Quick Plasmid Miniprep Kit, Invitrogen Corp., Carlsbad, CA.
k.
MWG Biotech, Ebersberg, Germany.
l.
TaqMan Universal PCR Master Mix, Applied Biosystems, Foster City, CA.