
Abstract
A chronic progressive neurological condition in an Alexandrine parrot (Psittacula eupatria) was manifest as intention tremors, incoordination, and seizure activity. Histology revealed large eosinophilic bodies throughout the central nervous system, and electron microscopy demonstrated that these bodies were greatly expanded axons distended by short filamentous structures that aggregated to form long strands. The presence of periodic acid–Schiff-positive material within the neuronal bodies of Purkinje cells and ganglionic neurons is another distinctive feature of this disease. The histological features of this case display some features consistent with giant axonal neuropathy as reported in humans and dogs. Based on investigation of the lineage in this case, an underlying inherited defect is suspected, but some additional factor appears to have altered the specific disease presentation in this bird.
Reports of neurodegenerative diseases in birds are rare. Lafora disease has been described in cockatiels, 4 and cerebellar degeneration has been noted in juvenile parrots 18 and chickens, 17 the latter as an inherited sex-linked disorder. Lysosomal storage disease with neurological involvement has also been diagnosed in emus 15 and Humboldt penguins. 22 A number of nutritional deficiencies may induce neurological degeneration in birds, in particular encephalomalacia associated with vitamin E deficiency, 5 and peripheral neuropathy and myelinopathy caused by riboflavin deficiency. 14 Axonopathies in birds most commonly result from exposure to toxins such as organophosphates and heavy metals, although a hereditary axonopathy characterized by reduced axonal size has been documented in neurofilament-deficient quail. 22
Giant axonal neuropathy is a rare condition caused by a heterogeneous range of recessive defects in the gigaxonin (GAN) gene. 3 Gigaxonin regulates neurofilament degradation by the ubiquitin–proteasome pathway, and defects in gigaxonin expression lead to accumulation of neurofilaments within axons. 16 The condition has been reported in humans and dogs as an inherited defect,1,10 and has been induced experimentally in mice. 8 The disease manifests as early-onset motor and sensory neuropathy that progresses to central nervous system signs with seizures and eventual death.
A 21-month-old male hand-reared Alexandrine parrot was presented to a veterinary hospital for investigation of longstanding and progressive weight loss, lethargy, and incoordination causing the bird to fall from its perch. On examination, the bird displayed a generalized intention tremor that was demonstrable with step-up and step-down exercises. The tremors were not present at rest, and no spontaneous nystagmus was observed. The bird was alert, and appeared to comprehend and respond to the owner’s commands. Clinical examination was otherwise unremarkable. Hematological and biochemical findings from blood samples obtained at multiple ages are presented in Table 1. Persistent and fluctuating increases in glutamate dehydrogenase and aspartate aminotransferase were noted, together with a mild monocytosis and basophilia. Based on the enzyme elevations, hepatocellular damage was suspected, and a liver biopsy was performed. The biopsy specimen was reported to display mild biliary hyperplasia and fibrosis, and a single small focus of necrosis was also observed. Periodic acid–Schiff (PAS) staining of the biopsy specimen did not indicate the presence of a carbohydrate storage disease.
Hematological and biochemical results from blood samples obtained at different ages.*
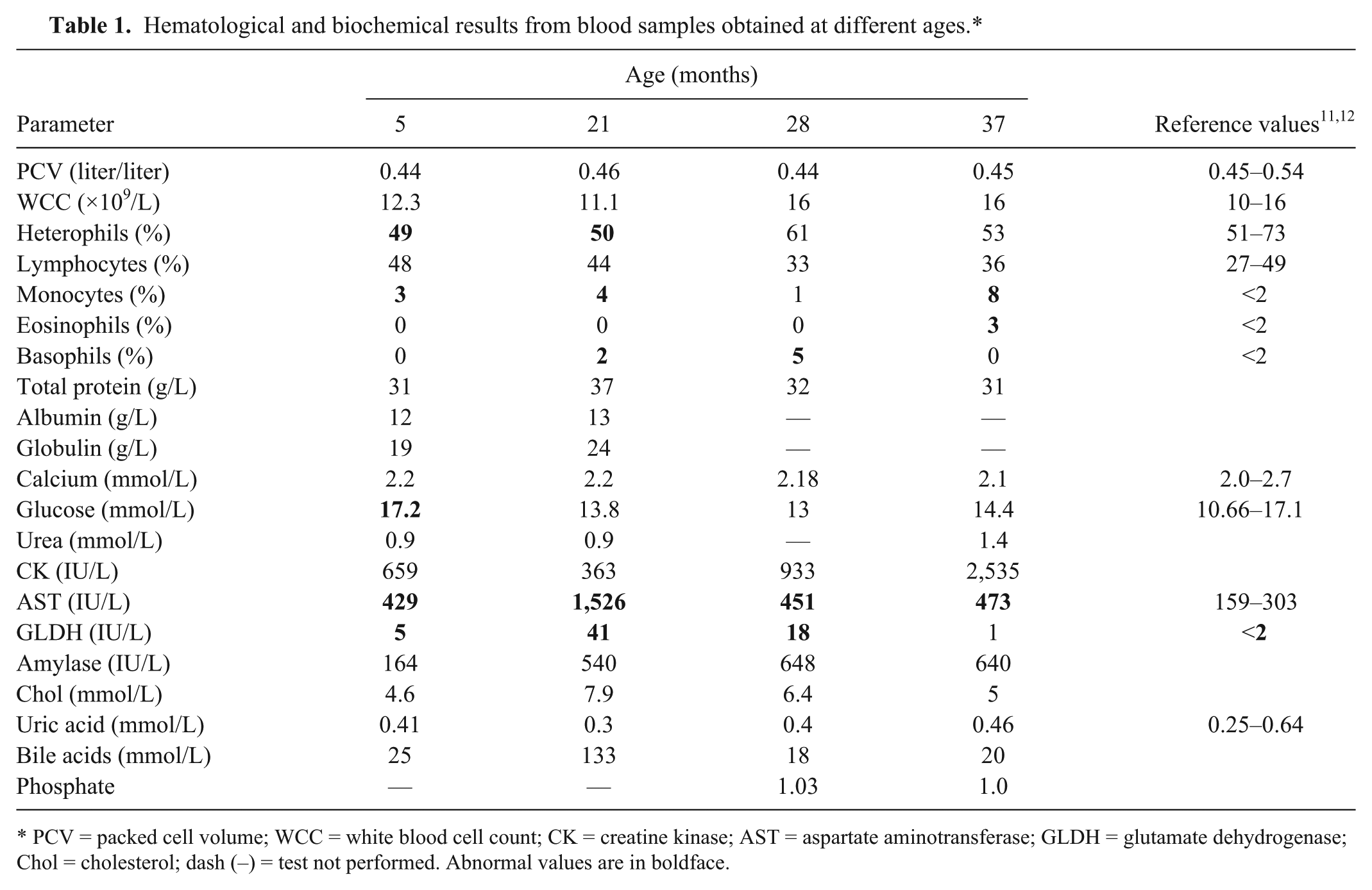
PCV = packed cell volume; WCC = white blood cell count; CK = creatine kinase; AST = aspartate aminotransferase; GLDH = glutamate dehydrogenase; Chol = cholesterol; dash (–) = test not performed. Abnormal values are in boldface.
Serological testing for Toxoplasma using a modified agglutination test 9 was negative. Polymerase chain reaction (PCR) testing for Chlamydophila 19 performed on a mixed conjunctival, choanal, and cloacal swab was also negative, as was PCR for avian polyomavirus performed on blood. PCR for Beak and feather disease virus was performed by the referring veterinarian and was reported to be negative.
The clinical signs failed to improve following treatment with doxycycline a (50 mg/kg, pectoral intramuscular injection, once weekly) or itraconazole b (3.5 mg/kg orally, once daily), and the intention tremor progressively worsened, with development of pressure sores on both hocks because of impaired mobility. By 3 years of age, the bird was also displaying frequent seizures. The clinical signs partially responded to a trial of phenobarbitone c (1 mg/kg orally, twice daily as ongoing therapy), with reduction in both tremor severity and seizure frequency, but 1 month later, at 39 months of age, the bird died suddenly with nasal and oral hemorrhage.
No gross lesions were noted at autopsy; the brain and samples of liver, heart, lung, gizzard, duodenum, pancreas, spleen, adrenal, kidney, eye, and skeletal muscle were fixed in 10% buffered formalin for histological examination. Histologically within the brain, affecting both gray and white matter, there were widely distributed, variably sized (up to ~100 µm in diameter), round to elongate, well-defined eosinophilic structures that occasionally contained fine boat-shaped clefts, imparting a crinkled appearance (Fig. 1). The structures were particularly prominent within the cerebellar nuclei and arbor vitae, superficial optic tectum, and midbrain, and were sometimes associated with vacuolation adjacent to or within the eosinophilic bodies (Fig. 2). Occasionally, swollen axons were visible within peripheral nerves (Fig. 3).
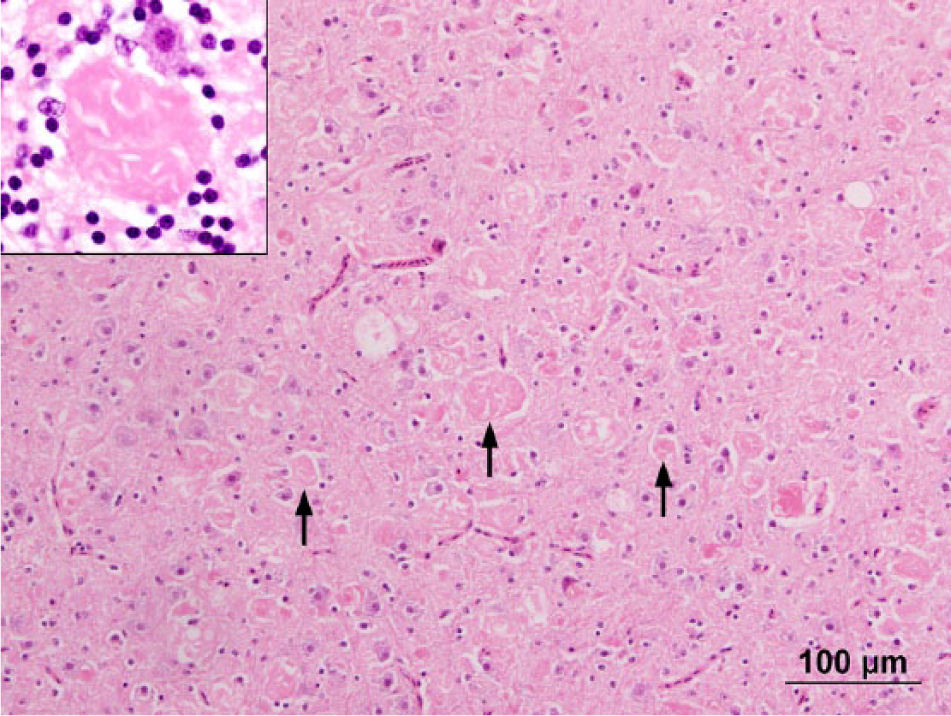
Thalamus. Variably sized eosinophilic bodies (arrows) are present throughout the tissue. Inset shows detail of eosinophilic body displaying clear clefting, with an adjacent neuron. Hematoxylin and eosin. Bar = 100 μm.
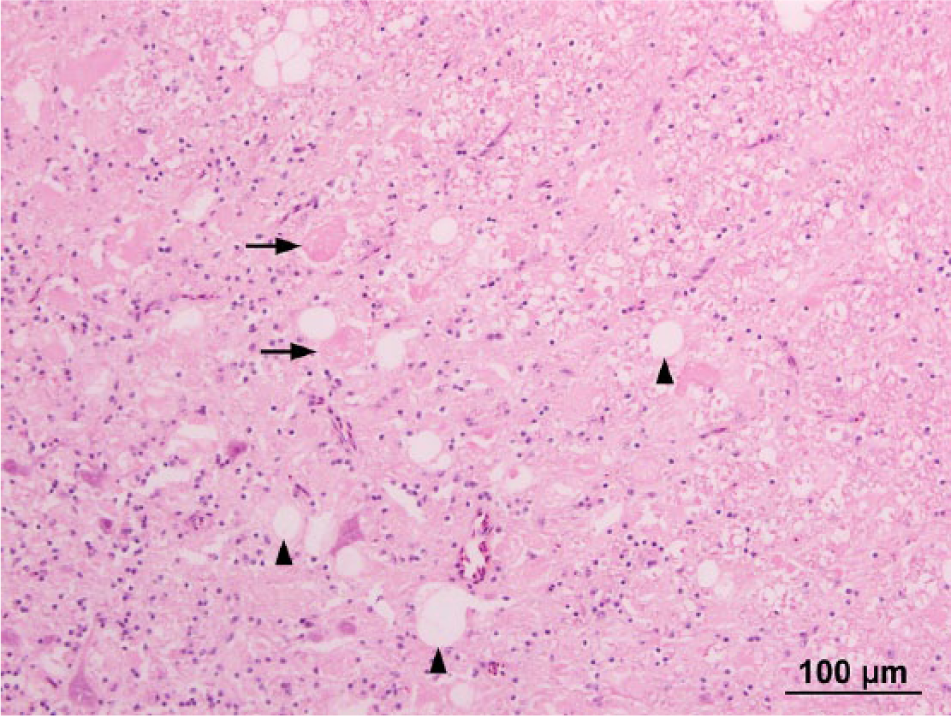
Cerebellar nuclei. Eosinophilic bodies (arrows) are interspersed by variably sized vacuoles (arrowheads). Hematoxylin and eosin. Bar = 100 μm.
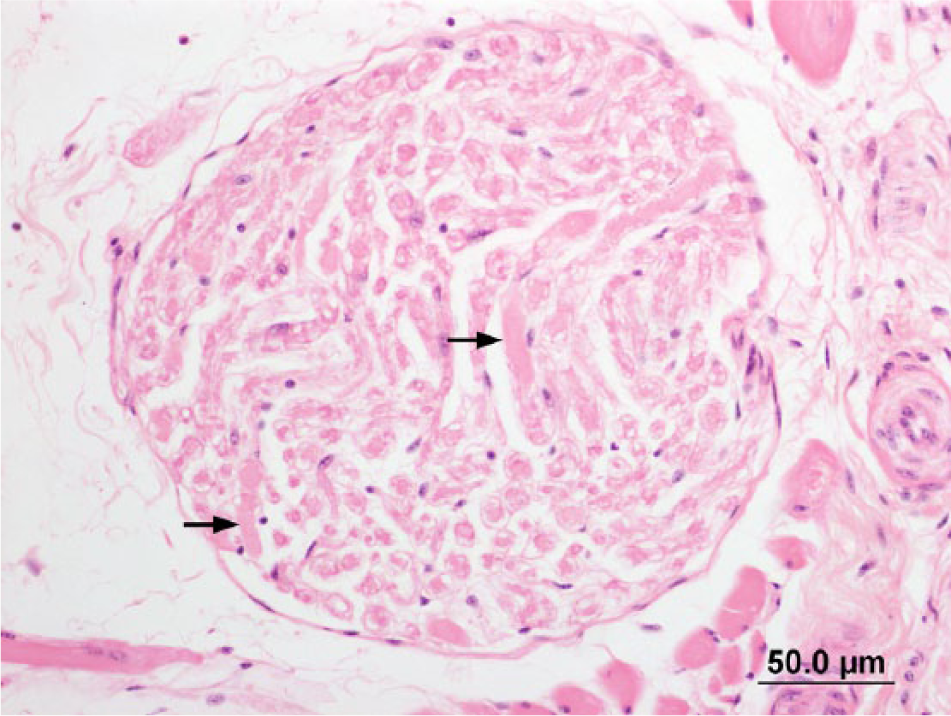
Peripheral nerve. Occasional swollen eosinophilic axons (arrows) are visible within the nerve. Hematoxylin and eosin. Bar = 50.0 μm.
Hepatocytes displayed mild swelling with finely granular cytoplasm, consistent with recent phenobarbitone therapy. Changes consistent with the previous liver biopsy findings were not observed, and a cause for the persistent elevation of liver-associated enzymes in serum was not identified. Fresh hemorrhage was present within the bronchi and parabronchi, suggestive of acute aspiration, and head trauma with aspiration of hemorrhage was proposed as a cause of death.
The eosinophilic bodies within the brain did not stain positively with Congo red, Luxol fast blue, PAS, Bodian, or Gallyas stains. Large accumulations of PAS-positive material were present in some cerebellar Purkinje cells (Fig. 4), and multiple small round bodies composed of similar material were noted within pericardial ganglia. Immunohistochemistry performed with an anti-neurofilament antibody d failed to stain the eosinophilic bodies, but the axonal swellings within the peripheral nerves were highlighted, and scattered neurofilament-positive axonal swellings were also identified within the brain.
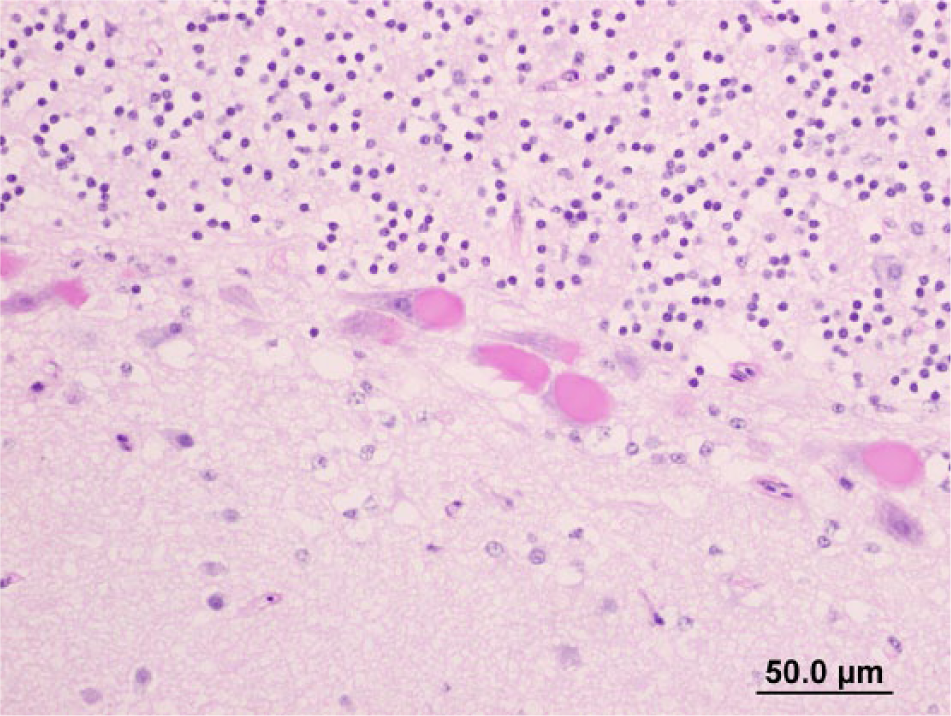
Cerebellum. Periodic acid–Schiff-positive bodies within Purkinje cells. Bar = 50.0 μm.
Electron microscopy imaging identified the eosinophilic material as accumulations of short filaments within massively expanded axons (Fig. 5); these filaments often aggregated in long strands and were occasionally interspersed by clear clefts or vacuoles (Fig. 6). The material was largely confined to axons, and accumulation within the neuronal body was minimal. Both myelinated and unmyelinated axons were affected, although myelin sheaths surrounding distended axons were often thin. Although occasional mitochondria were present within the aggregates of material, the majority of organelles were displaced and compressed by the accumulations.
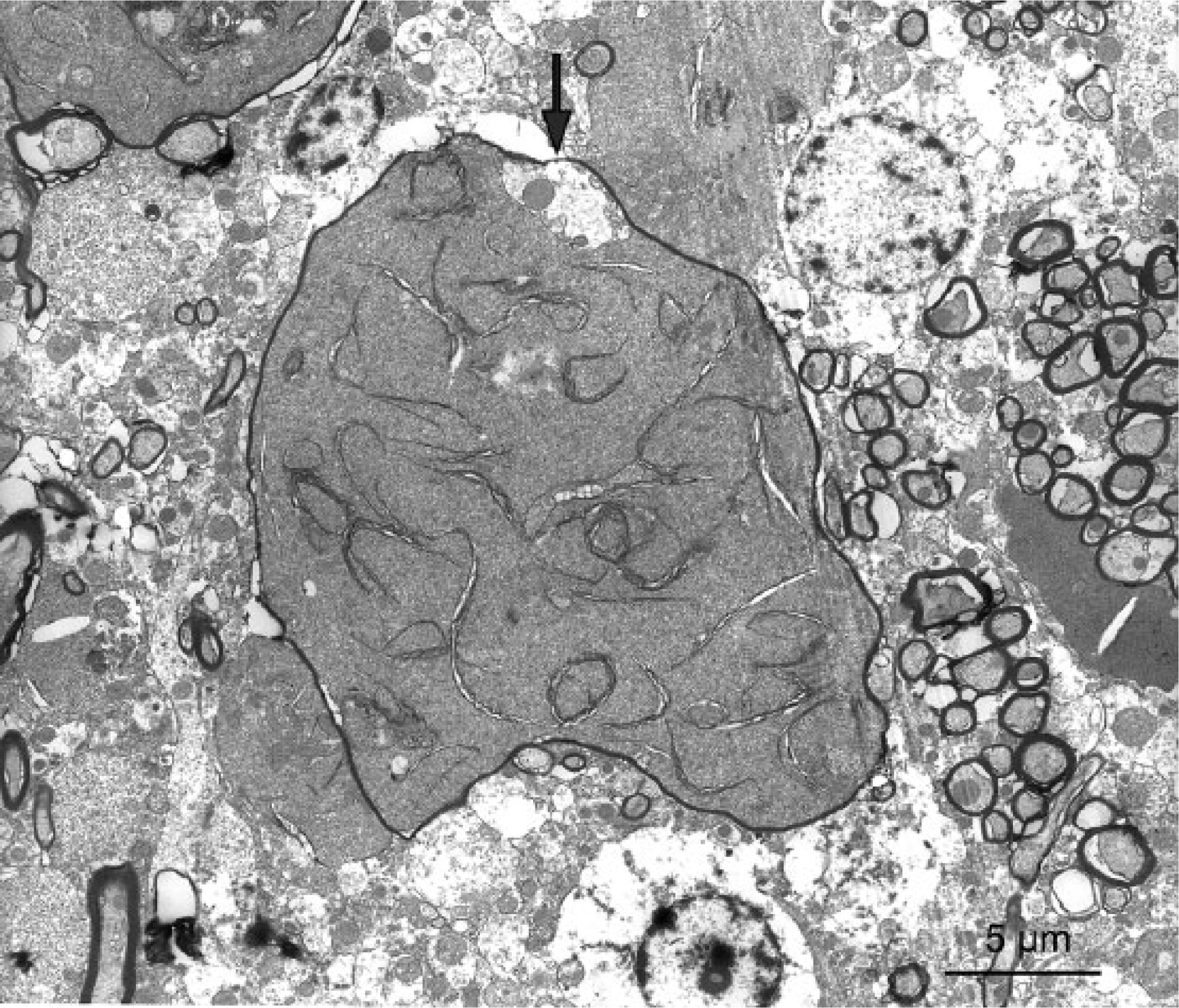
Brain. Free filaments and long compacted strands greatly distend a thinly myelinated axon. Compare to the size of adjacent normal myelinated axons. Note the small, pale surviving pocket of cytoplasm with organelles (arrow). Electron microscopy, 3,800×. Bar = 5 μm.
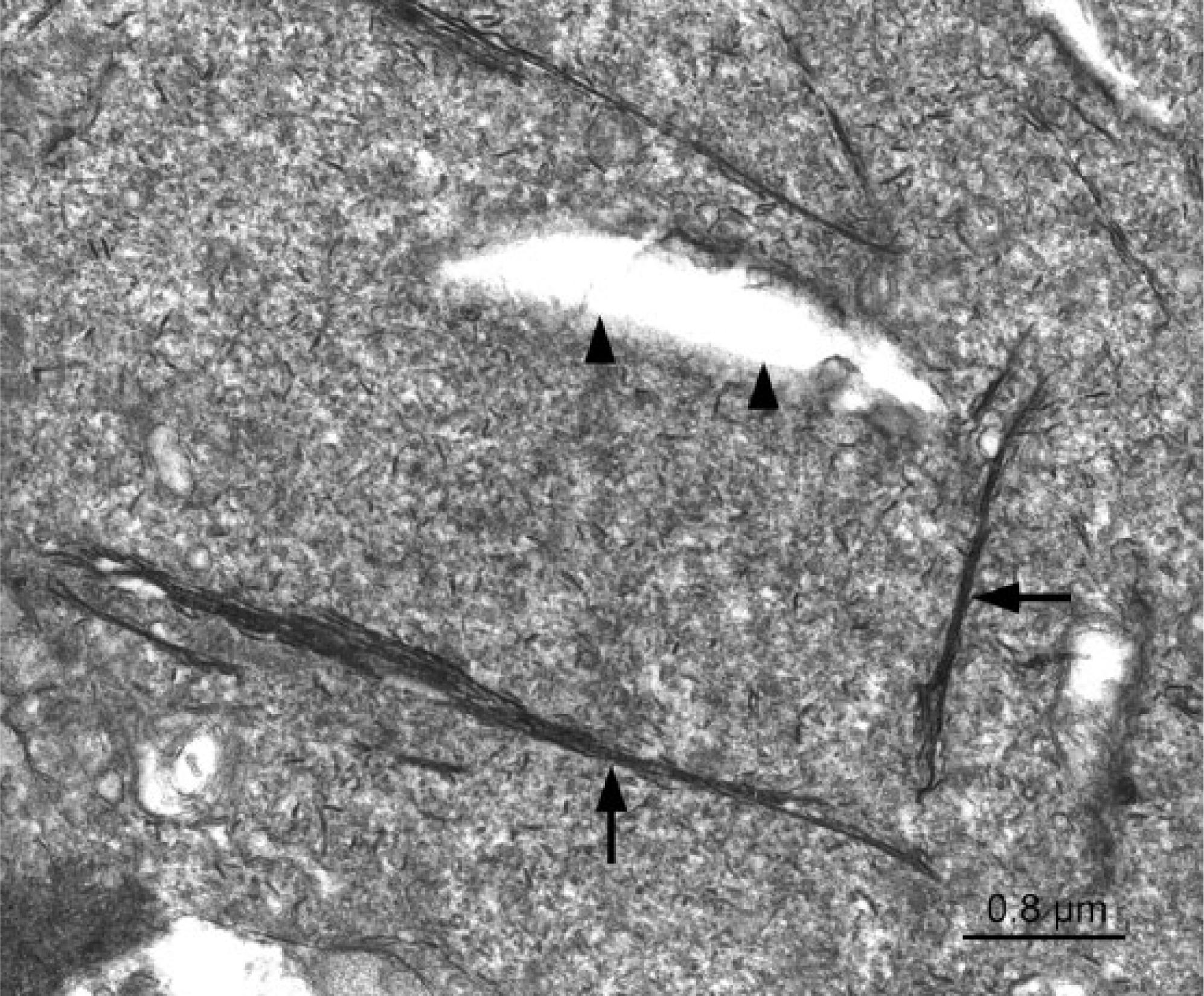
Brain. Short filamentous structures aggregating to form long strands (arrows). A clear cleft in the material is also present (arrowheads). Electron microscopy, 25,600×. Bar = 0.8 μm.
The short filamentous nature of the deposited material in this case—together with its propensity to form long strands—is most consistent with a cytoskeletal element, and the localization of the material within axons is suggestive of a form of neurofilament. However, the absence of neurofilament positivity with immunohistochemistry, together with the negative Bodian staining, suggests that the material does not contain any of the normal major subunits associated with neurofilament triplet proteins in mammals and birds. 2 This is distinct from the human form of giant axonal neuropathy, in which the axonal aggregates are both argyrophilic and neurofilament positive.1,20 The lack of staining with Gallyas stain further indicates that the aggregates do not contain tau protein, 13 a microtubule-associated protein. Although formalin fixation precluded further identification of the material, it is possible that the bodies may represent an accumulation of other structural elements of neurons, such as internexin, nestin, peripherin, or β-tubulin III. While it is possible that the neurofilament-positive axonal swellings could represent an earlier stage of the disease wherein the eosinophilic bodies retain neurofilament immunoreactivity, it is more likely that they reflect secondary axonal degeneration with accumulation of neurofilaments.
The present case also displayed several other features that were inconsistent with classical giant axonal neuropathy. First, the long filamentous aggregates and clefts observed on electron microscopy have not been reported with giant axonal neuropathy in other species, which more typically display accumulation of fine homogeneous neurofilaments that occasionally form a faint whorled pattern. 1 The appearance of the clefts within the material is suggestive of cholesterol, but could also represent artefactual change.
Second, a distinctive histological feature observed in this bird was rare deposition of PAS-positive material within the soma of Purkinje cells, with similar material also noted within ganglial neuronal bodies in the pericardium. While Lafora disease has been reported in psittacines with a similar clinical presentation, 4 the intraneuronal bodies in this bird displayed a hyaline appearance, were irregular in shape, and lacked the typical central core of Lafora bodies. The scarcity of these bodies prevented identification on electron microscopy, but the distinct localization and staining characteristics indicate either that posttranslational glycosylation of the accumulating filamentous material is occurring within these cells, or that there is abnormal accumulation of multiple types of material within the central nervous system. If the latter, it remains unclear whether this reflects a primary transport defect, or whether the PAS-positive material accumulates secondary to axonal obstruction.
Third, in other species, giant axonal neuropathy is typically (though not necessarily) associated with curly hair10,21; no gross feather abnormalities were identified in this case. For these reasons, we believe that this condition is distinct from classical giant axonal neuropathy as reported in other species, and may represent a novel disease process.
Investigation into the lineage of this bird revealed a familial history of neurological disease, with affected parrots developing nonprogressive neurological signs at ~2 months of age. Affected birds displayed opisthotonus when stressed and circled to the ground when flight was attempted. This trait displayed an autosomal dominant pattern of inheritance, and the bird in the present report was the offspring of an affected hen and unaffected cock. However, despite the evidence in favor of an inherited condition, the distinct clinical presentation of the case described herein (progressive tremors and seizures) suggests another factor may be influencing the disease course in this instance.
Aside from degenerative disease, a toxic etiology was also considered, particularly as the biochemistry in this case was suggestive of a hepatopathy. Several toxins are known to induce axonal swellings similar to those observed in giant axonal neuropathy, including acrylamide, iminodipropionitrile, and hexacarbons.6,7 However, toxin exposure was unlikely in this case, as the chemicals involved are uncommon outside of an industrial or laboratory setting, and the bird was hand reared and housed in a controlled residential environment. This bird was also reported to have displayed neurological abnormalities from a young age with gradual progression over several years, which is more consistent with a congenital defect. Moreover, the aggregated strands noted ultrastructurally in this bird have not been reported previously in cases of toxic axonopathy.
Footnotes
Acknowledgements
We acknowledge Dr. Cedric Raine (Albert Einstein College of Medicine, Bronx, New York) for assistance with the ultrastructural interpretation, and Faye Docherty and Paul Benham for their assistance with histological processing.
Authors’ contributions
A Stent contributed to acquisition, analysis, and interpretation of data, and drafted the manuscript. M Gosbell contributed to acquisition of data. L Tatarczuch contributed to acquisition and interpretation of data. BA Summers contributed to interpretation of data. M Gosbell, L Tatarczuch, and BA Summers critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
Psittavet Injection, Vetafarm Pty. Ltd., Wagga Wagga, New South Wales, Australia.
b.
Itraconazole, Bova compounding, Caringbah, New South Wales, Australia.
c.
Phenomav, Mavlab, Slacks Creek, Queensland, Australia.
d.
Monoclonal mouse anti-human neurofilament protein (clone 2F11), Dako Denmark A/S, Glostrup, Denmark.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.