
Abstract
Agamid adenovirus 1 (AgAdv-1) is a significant cause of disease in bearded dragons (Pogona sp.). Clinical manifestations of AgAdv-1 infection are variable and often nonspecific; the manifestations range from lethargy, weight loss, and inappetence, to severe enteritis, hepatitis, and sudden death. Currently, diagnosis of AgAdv-1 infection is achieved through a single published method: standard nested polymerase chain reaction (nPCR) and sequencing. Standard nPCR with sequencing provides reliable sensitivity, specificity, and validation of PCR products. However, this process is comparatively expensive, laborious, and slow. Probe hybridization, as used in a TaqMan assay, represents the best option for validating PCR products aside from the time-consuming process of sequencing. This study developed a real-time PCR (qPCR) assay using a TaqMan probe–based assay, targeting a highly conserved region of the AgAdv-1 genome. Standard curves were generated, detection results were compared with the gold standard conventional PCR and sequencing assay, and limits of detection were determined. Additionally, the qPCR assay was run on samples known to be positive for AgAdv-1 and samples known to be positive for other adenoviruses. Based on the results of these evaluations, this assay allows for a less expensive, rapid, quantitative detection of AgAdv-1 in bearded dragons.
Adenoviruses are nonenveloped DNA viruses that range from 60 to 100 nm in diameter. Infections with adenoviruses are well documented, and have been reported in all of the bony vertebrates, including squamates, from which the genus Atadenovirus likely originated.3,5,19 Currently, there are 5 accepted genera of adenoviruses within the family Adenoviridae: Atadenovirus (predominantly in squamate hosts), Aviadenovirus (in avian hosts), Ichtadenovirus (isolated from a white sturgeon), Mastadenovirus (in mammalian hosts), and Siadenovirus (found in chelonian, amphibian, and avian species). Additionally, a sixth genus has been proposed (Testadenovirus), whose members were isolated from hosts in the order Testudines. 2 Many reptilian adenoviruses appear to have coevolved with the species they infect, leading to a fair amount of epidemiological species specificity.17–19
Bearded dragons (Pogona sp.) are agamid lizards native to Australia that have become an increasingly popular pet and are frequently found in zoological collections. Although exportation of these animals from Australia is illegal, a successful breeding industry has led to numerous animals in captivity. The most common species, the central bearded dragon (Pogona vitticeps), is represented by more than 700 individuals in 258 zoological institutions worldwide (International Species Information System. Species holdings. 2014. https://zims.isis.org). In general, bearded dragons make good pets, and typically thrive in captivity, although a number of diseases have been documented.
Agamid adenovirus 1 (AgAdV-1) appears to be a highly prevalent disease of concern in Pogona species. Adenoviral disease was first reported in 1982, 10 and there have been numerous documented cases of AgAdV-1 infections in bearded dragons.4,9,11 In 2004, the first specific characterization of AgAdV-1 was reported, 19 and it appears to be widespread in P. vitticeps, having been identified in numerous animals in North America, Europe, and Australia.1,6,12,15,18 It has also been identified in a central netted dragon (Ctenophorus nuchalis) in northern Australia. 6 Partial AgAdV-1 hexon gene sequences in bearded dragons in the United States demonstrated 4 different genotypes; diversity was not seen in the polymerase gene. 18 Only 1 other adenovirus has been reported from the genus Pogona, a helodermatid AdV-2–like virus that was identified in a P. minor. 6 Although there have been reports of subclinical adenoviral infection in reptiles,8,16 many infections result in moderate to severe clinical disease.
A myriad of clinical signs have been associated with AgAdV-1 infection, including lethargy, anorexia, dehydration, regurgitation, vomiting, diarrhea, central nervous system signs (e.g., paresis, head tilt, circling, and opisthotonos), hepatitis, enteritis, nephritis, and sudden death.12,15,17 Disease can occur at any age, but is seen more frequently in juveniles.
Gross pathologic lesions frequently involve the liver (i.e., hepatomegaly, mottled appearance, and pallor), 7 and luminal hemorrhage of the intestines. 13 Histologic identification of amphophilic intranuclear inclusions in hepatocytes and enterocytes is suggestive of infection.
To date, diagnosis of AgAdV-1 infection is achieved through a single published method: standard nested polymerase chain reaction (nPCR) and sequencing of sample material (e.g., fecal, cloacal wash, hepatic parenchyma). 19 This process involves extracting DNA from the sample, then running PCR using primers that are specifically designed to amplify adenoviral species. This is followed by agarose gel electrophoresis to separate the PCR products, extraction of appropriately sized bands from the gel, cycle sequencing of the extracted band of DNA, running a sequencing gel, and analysis of the data obtained from the sequence. Although sensitive and specific, this process is relatively expensive, laborious, and extremely time-intensive.
In contrast, hydrolysis probe real-time PCR (qPCR) uses hybridization of a sequence-specific probe during the PCR reaction to validate the product identity. As the polymerase advances past the hybridized probe, the probe is digested and releases labeled dyes. The dyes are then measured spectrophotometrically. Thus, the synthesis of specific PCR product is determined as the reaction progresses, providing not only qualitative information regarding presence but also quantitative information. The steps are significantly less labor-intensive, involving only the DNA extraction from the sample, qPCR, and simpler data analysis. In addition, qPCR assays have been used successfully for many infectious agents, and are often found to be more sensitive than standard PCR. This simpler, less-expensive assay provides the ability to perform cost-effective cross-sectional evaluations of collections that contain potentially susceptible animals, enabling management practices to help reduce morbidity and mortality in collections of Pogona sp.
The aim of this study was to develop a hydrolysis probe qPCR assay to diagnose the presence of AgAdV-1. This assay will provide a faster and less-expensive option than the option available to date, and will provide quantitative information.
The samples selected for AgAdV-1 analyses included 19 diagnostic samples from bearded dragons previously submitted to the authors’ laboratory. These samples had previously been confirmed positive for AgAdV-1 via standard PCR and sequencing. An additional 13 adenovirus PCR–negative samples from 10 central bearded dragons, 2 leopard geckos (Eublepharis macularius), and a Solomon Island skink (Corucia zebrata) were included as species-specific negative controls.
Twenty additional DNA extractions from clinical samples containing other adenoviruses were used to determine the specificity of the assay. These samples included snake adenoviruses 1 and 4, crotalid adenoviruses 1 and 2, helodermatid adenoviruses 1 and 2, murine adenovirus 2, Sulawesi tortoise adenovirus 1, gouldian finch adenovirus 1, pancake tortoise adenovirus 1, eastern box turtle adenovirus 1, and psittacine adenovirus 1.
To prepare the target standards, DNA from a known AgAdV-1–positive sample was amplified using nested degenerate primers designed to isolate a diverse range of adenoviruses. 19 The PCR product was resolved on a 1.5% agarose gel, extracted, and confirmed as AgAdV-1 by direct Sanger sequencing. The DNA concentration was determined with a spectrophotometer,a and the qPCR standard curve was generated by analysis of 10-fold serial dilutions ranging from 101 to 106 copies per well. A standard concentration of 106 copies per well was prepared and aliquoted for single use with each run. Aliquots were stored at -80°C until the day of use.
Quantitative PCR was performed on samples using the forward primer AgAdV-1-F (5′-CATGCCGTGCGGTCGTA-3′), reverse primer AgAdV-1-R (5′-TTGGAAGCGTCTGATTTCGA-3′), and probe targeting the AgAdV-1 conserved region (5′-[FAM]-CTTGCCGCCTTTGGACGCGT-[BHQ]-3′). The AgAdV-1–specific target region was selected by comparison with other aligned adenoviral polymerase sequences.
Each 20-μL qPCR reaction was run in duplicate using a standard fast protocol. Each reaction consisted of 10 µL of TaqMan master mix,b 1 µL each of AgAdV-1–specific primers and probe (12 µM AgAdV-1-F and AgAdV-1-R primers [600 nM/reaction], 5 µM AgAdV-1 probe [250 nM/reaction]), and 3 µL of nuclease-free water. A total of 16 µL of the reaction mix and 4 µL of samples were added into 96-well reaction plates.c Nuclease-free water was added to 6 wells of each plate to serve as non-template controls. A eukaryotic 18S rRNA endogenous control kitd was used to validate the presence of amplifiable DNA in each sample in a separate well. The standard curve used 10-fold serial dilutions of the AgAdV-1 PCR amplicon from the index case. The template was quantified by both spectrophotometry and comparison to gel standards. Dilutions were made, and control standards were run using 101–106 copies per well in duplicate on each plate. The quality of the qPCR probe and primers were assessed by evaluating the overall efficiency of the reaction and the linear confidence (R2) of the curve. Reactions were amplifiede with cycling conditions as follows: initial denaturation at 95°C for 20 sec, followed by 45 amplification cycles of 95°C for 3 sec and 60°C for 30 sec.
The standard curve efficiency was evaluated by analyzing 10-fold serial dilutions (1:10) of the AgAdV-1 polymerase amplicon ranging from 106 copies/reaction to 101 copies/reaction. These dilutions were assayed in triplicate on 4 plates. Acceptable efficiencies were defined by a target range between 80% and 110%, with a linear confidence (R2) >0.98.
To evaluate the diagnostic sensitivity and specificity, the qPCR results were compared to the gold standard nPCR. The analytic sensitivity of the assay was determined by analyzing 2-fold serial dilutions (1:2) of the AgAdV-1 polymerase amplicon to determine the lowest count/well value that consistently yielded replicates with curves above the threshold (i.e., the limit of detection). The analytic specificity of this assay was assessed by performing the qPCR with the adenovirus-positive DNA samples of other adenoviral species, as mentioned above.
The standard curve generated by 10-fold serial dilutions of DNA was found to be linear, with a slope ranging from 3.60 to 3.88, indicating 89.57% to 81.02% efficiency and an R2 value ranging from 0.9834 to 0.9989 (Fig. 1). The threshold cycle values ranged from 22.1 cycles at 106 copies to 40.8 cycles for 10 copies. Samples run on the qPCR were compared with the standard curve to estimate the number of copies present, and results are given in Table 1.
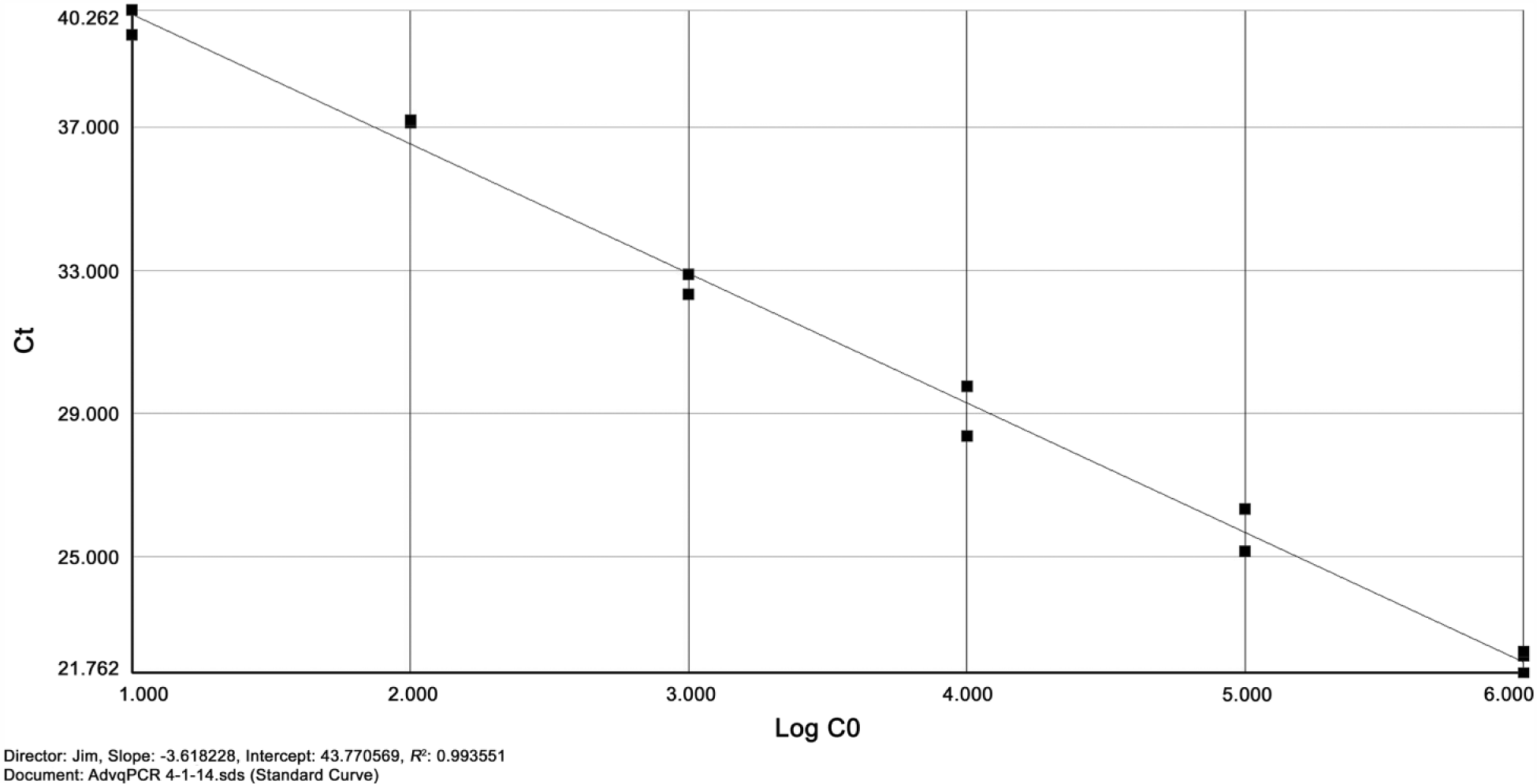
Standard curve of agamid adenovirus 1 real-time polymerase chain reaction. Vertical axis shows threshold cycle (Ct); horizontal axis shows number of copies present on a logarithmic scale.
Results of probe hydrolysis real-time polymerase chain reaction (qPCR) assay for agamid adenovirus 1 (AgAdv-1) in comparison to the gold standard (PCR and sequencing).
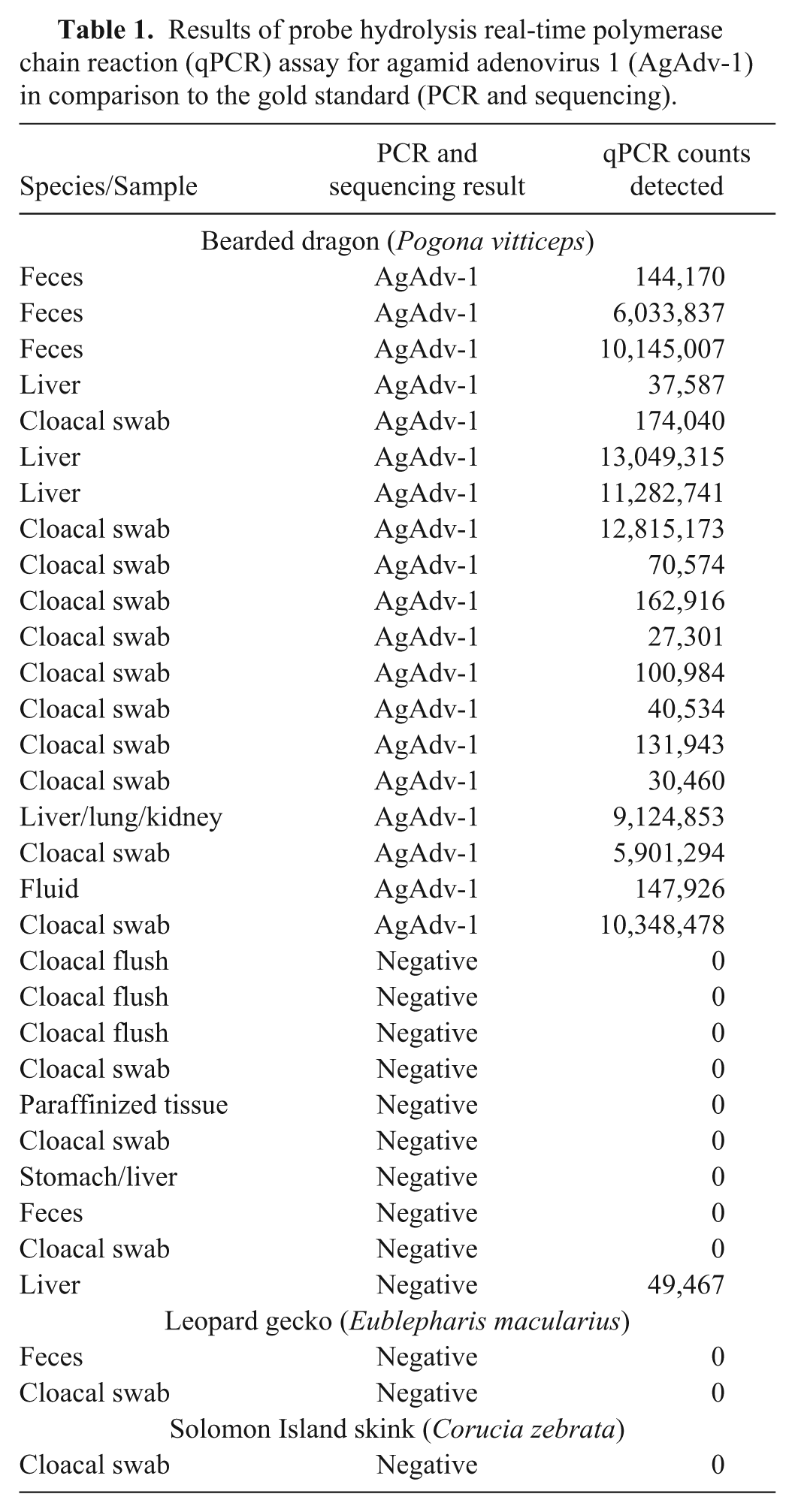
The linear dynamic range for this qPCR assay was between 10 and 1 × 106 copies. The assay was able to reliably detect 10 copies per well. None of the 20 additional non–AgAdV-1 adenovirus positive samples tested positive in the qPCR assay for AgAdV-1.
When compared with the current gold standard test (PCR and sequencing), the assay developed in this study correctly identified all 19 positive control samples as positive for AgAdV-1. Additionally, this assay provided quantitative data regarding the number of DNA copies in a given positive sample. Comparison to the gold standard for evaluation of negative controls resulted in 12 negative results using the qPCR assay. A single sample, which had been identified as negative with standard PCR, was identified as positive using the qPCR assay. To determine whether this was a false positive in the qPCR or a false negative in the standard PCR, a third nPCR protocol was run on the discordant sample. This first utilized the inner primers of the standard PCR as a first round (polFinner and polRinner), and was followed by running 2 different second rounds involving the qPCR primers (polFinner with AgAdV-1-R, and AgAdV-1-F with polRinner). The products were then sequenced and identified as AgAdV-1.
The results of this study validate and confirm the successful development of a qPCR assay to detect and quantify copies of AgAdV-1 in clinical diagnostic samples. With this development, the presence of AgAdV-1 in samples from Pogona sp. can be identified in a matter of hours rather than the several days that are typical using standard PCR and sequencing. This reduces turnaround time for practitioners awaiting results, and mitigates financial cost to the client, clinician, and the laboratory.
This assay successfully detected AgAdV-1 with as few as 10 copies of target DNA in a sample, suggesting very good analytic sensitivity. In addition, the analytic specificity for this assay appears to be excellent, with no non–AgAdV-1 adenovirus positive samples testing falsely positive.
In comparison with the gold standard (i.e., standard PCR with sequencing), the qPCR assay detected 100% of the AgAdV-1–positive controls, suggesting extremely good diagnostic sensitivity. However, while testing known negative samples, the qPCR assay identified 1 of the 13 samples as positive, yielding 92.3% diagnostic specificity. This sample, previously identified as negative by standard PCR, was quantified by qPCR to have more than 49,000 copies of target DNA. Quantification of positive samples revealed a range from 27,000 to more than 13 million copies of target DNA. In light of this, 49,000 copies represented a comparatively low count. Although this would appear to negatively impact the qPCR assay’s specificity, this result should be interpreted with caution in light of the limitations of using the standard PCR as the benchmark for diagnostic comparison. In the interest of clarifying the cause of this discrepancy, the additional PCR protocol was run. The result of this third PCR protocol confirms that this was a false-negative result from the gold standard PCR. Indeed, the data obtained through this study suggests that the qPCR assay may in fact be more sensitive than the gold standard PCR. Counts of positive samples were surprisingly high, with 8 above the range of the standard curve.
It should be noted that these qPCR values are for copies of target DNA detected, which may potentially differ from actual copy numbers and organisms present. Culture of AgAdV-1 was not possible, and thus we were unable to test pure organism samples directly. Instead, dilutions of known copy numbers of AgAdV-1 PCR amplicon were used as a standard curve. This control is a DNA template and does not reflect loss during extraction. The presence of PCR inhibitors or nucleases may result in falsely low readings. These are common in feces, so this is of special concern for fecal samples, which are commonly used for antemortem testing. 14 Furthermore, extraction efficiency may differ between tissue samples and fecal samples, so caution should be used to avoid over-interpretation when comparing different sample types.
The findings of this study have multiple ramifications. The first and most immediate is the development of a test that can accurately detect the presence of AgAdV-1 and also quantify the viral load in a given sample. Currently, the distribution of viral load in an infected bearded dragon has not been well evaluated. The range of clinical signs associated with infection suggests variation in site of viral pathology, and the reason for this is unclear. The ability to quantify the number of copies in a given sample would aid in understanding the burden of infection in different tissues, and may better elucidate the pathogenesis of disease.
With this qPCR assay, clinical samples can also be processed rapidly, giving clinicians valuable answers and allowing them to act on the information expeditiously. Because infection with AgAdV-1 can cause severe disease, and has the potential to devastate collections of Pogona sp., rapid response to clinical signs may be crucial to limiting additional morbidity and mortality. This includes decisions made regarding additions of new animals to collections. With a rapid and cost-effective test, groups of bearded dragons, either in zoological settings or in private collections, can be screened routinely.
An additional potential benefit of this assay is the ability to rapidly screen samples from wild populations. Little is known regarding the prevalence of AgAdV-1 in free-ranging individuals. The use of this assay would allow field researchers to collect samples from the wild and cost-effectively test them to provide data regarding the distribution of AgAdV-1 in noncaptive individuals.
In this report, we describe the development of a qPCR assay that provides a rapid, cost-effective, sensitive, and specific method for detecting AgAdV-1 in a given sample. We hope this assay will be of use for clinical evaluation, further surveillance of the disease, epidemiological studies, population management, and disease distribution.
Footnotes
a.
NanoDrop, Thermo Fisher Scientific Inc., Waltham, MA.
b.
TaqMan Fast universal PCR master mix 2× No AmpErase UNG, Life Technologies, Grand Island, NY.
c.
MicroAmp Fast optical reaction plates, Applied Biosystems, Foster City, CA.
d.
VIC/MGB probe, Life Technologies, Grand Island, NY.
e.
7500 Fast real-time PCR system, Applied Biosystems, Foster City, CA.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) declared that they received no financial support for their research and/or authorship of this article.