
Abstract
Leptospirosis is caused by several pathogenic Leptospira species, and is an important infectious disease of dogs. Early detection of infection is crucial for an effective antibiotic treatment of the disease. Though different polymerase chain reaction (PCR) assays have been developed for detection of pathogenic Leptospira spp., thorough evaluation of the performance of these assays using dog urine samples has not been carried out. In the current study, the performance of 3 real-time PCR (qPCR) assays was assessed, 1 targeting the 16S ribosomal RNA (rRNA) gene and the other 2 targeting the lipL32 gene, a gene for the LipL32 outer membrane protein. With DNA extracted from laboratory-cultured pathogenic Leptospira spp., all 3 qPCR assays showed 100% specificity and had identical lower limits of detection. Compared to a conventional, gel-based PCR assay, all 3 qPCR assays were 100-fold more sensitive. There was a 100% agreement in the results of the 3 assays when tested on urine samples collected aseptically from 30 dogs suspected for leptospirosis. However, when tested on 30 urine samples that were collected by the free-catch method, the 16S rRNA–based assay falsely detected 13.3% of the samples as positive for pathogenic Leptospira spp. Nucleotide sequence analysis of the amplified DNA fragments showed that the assay resulted in false positives because of unrelated bacteria. All urine samples collected from 100 apparently healthy dogs at a local animal shelter tested negative for pathogenic Leptospira spp. These results highlight the importance of sample-specific validation of PCR-based diagnostic assays and the application of appropriately validated assays for more reliable pathogen detection.
Introduction
Leptospirosis, a zoonotic disease of global public health importance, is caused by spirochetes of the genus Leptospira. 9 Several domestic animals are susceptible to leptospirosis. 1 Rodents and some other animals can act as reservoirs of leptospires by developing asymptomatic carrier state of infection in their kidneys and excreting the bacteria in urine. 9 Human beings usually acquire the infection through exposure of mucosa or skin abrasions to water contaminated by urine from infected animals. 1
Members of the genus Leptospira include 13 pathogenic species and 6 noninfectious saprophytic species. 1 Based on differences in serological reactivity to the carbohydrate antigens of the lipopolysaccharide, pathogenic leptospires are classified into more than 260 serovars. 1 Leptospirosis in dogs is mostly caused by serovars within species L. interrogans and L. kirschneri. 5 The infected dogs show highly variable clinical signs, ranging from no clinical signs to severe life-threating kidney, liver, or pulmonary failure. 5 Vaccination can help prevent infection, but vaccinated dogs may still be susceptible to infection with other serovars that are not included in the vaccines. 19 Commercial vaccines currently available in the United States contain only 4 serovars (Canicola, Grippotyphosa, Icterohaemorrhagiae, and Pomona). Treatment with antibiotics is effective, but initiating the treatment within the first 7 days of infection is extremely important for a successful outcome. For example, a study in human beings suggested that antibiotic treatment after the initial 7 days of infection shows limited benefit in the face of severe clinical disease, making early detection of the disease critical. 3
The microscopic agglutination test (MAT) that detects the presence of serovar-specific antibodies in patient serum is commonly used for the diagnosis of Leptospira infection in dogs. 7 However, there are many caveats for the application of this test in diagnosis of canine leptospirosis, including negative results in acute infections, false-positive interpretation of the test results due to prior vaccination, conflicting results from different laboratories, and the limited number of serovars routinely used in the assay. 12 Polymerase chain reaction (PCR)-based assays are becoming the diagnostic tests of choice for rapid detection of infectious agents in clinical samples. However, while there are several reports of conventional and real-time PCR (qPCR) assays for the detection of pathogenic Leptospira spp., there is limited published information regarding the validation of these assays using canine urine samples.8,14
In the current study, 3 qPCR assays and 1 gel-based conventional PCR assay were compared for their ability to detect pathogenic Leptospira species in canine urine samples. Two of the evaluated qPCR assays, termed the Animal Disease Diagnostic Laboratory qPCR (ADDL qPCR) and the Centers for Diseases Control and Prevention qPCR (CDC qPCR) targeted the lipl32 gene encoding LipL32, a lipoprotein present only in the outer membrane of pathogenic Leptospira spp. 17 The ADDL qPCR was developed in the current study while the CDC qPCR has been previously described. 17 The third qPCR assay targeted the 16S ribosomal RNA (rRNA) gene (termed the 16S rRNA qPCR), and the primers and probe of this assay were reported to be specific to pathogenic Leptospira spp. 16 The traditional gel-based PCR assay utilized 2 sets of primers meant to be specific amplification from all pathogenic Leptospira spp. 6
Materials and methods
Primers and probes
Nucleotide sequences of the primers and probes used in this study are shown in Table 1. The primers and probe for the ADDL qPCR assay were designed using commercial software a to target a conserved region of the lipL32 gene of Leptospira strains. The CDC and ADDL qPCR assays target the lipl32 gene; positive amplification in the CDC and ADDL qPCR assays results in a 241-bp 17 and 76-bp long DNA fragment, respectively. The 16S rRNA qPCR assay amplifies an 88-bp fragment. 16 All of the primers and probes were custom synthesized at a commercial manufacturer. b All 3 probes contained 6-carboxyfluorescein (6-FAM) at the 5′-end as the reporter dye and black hole quencher 1 at the 3′-end as the quencher.
Primers and probes used in the current study.*

PCR = polymerase chain reaction; CDC = Centers for Disease Control and Prevention, Atlanta, Georgia; ADDL = Animal Disease Diagnostic Laboratory, West Lafayette, Indiana; 16S rRNA = 16S ribosomal RNA.
Real-time PCR assays
The assays were performed using Taq DNA polymerase c and a commercial thermocycler. d The 3 assays were individually optimized for Mg2+, deoxynucleotide triphosphates, primers and probe concentrations, and for the annealing temperature. The components of the final reaction mixture of these PCR assays are given in Table 2. The PCR cycling parameters for the CDC qPCR were 10 min at 95°C followed by 45 cycles of each consisting of 15 sec of denaturation at 95°C, 30 sec of annealing at 58°C, and 5 sec of extension at 72°C. For the ADDL qPCR, parameters of 10 min at 95°C followed by 45 cycles of each consisting of 15 sec of denaturation at 95°C, 30 sec of annealing at 62°C, and 5 sec of extension at 72°C were used. The 16S rRNA qPCR cycling parameters were 10 min at 95°C followed by 45 cycles of each consisting of 15 sec of denaturation at 95°C, 30 sec of annealing at 62°C, and 10 sec of extension at 72°C. The fluorescent data was acquired during the annealing step and analyzed by using the software on board the thermocycler. d The threshold cycle (Ct) values were determined by primary curve analysis program with the threshold limit set at 30 fluorescent units.
Concentrations of components in the final reaction mixtures of the 3 real-time polymerase chain reaction (qPCR) assays used in the current study.*
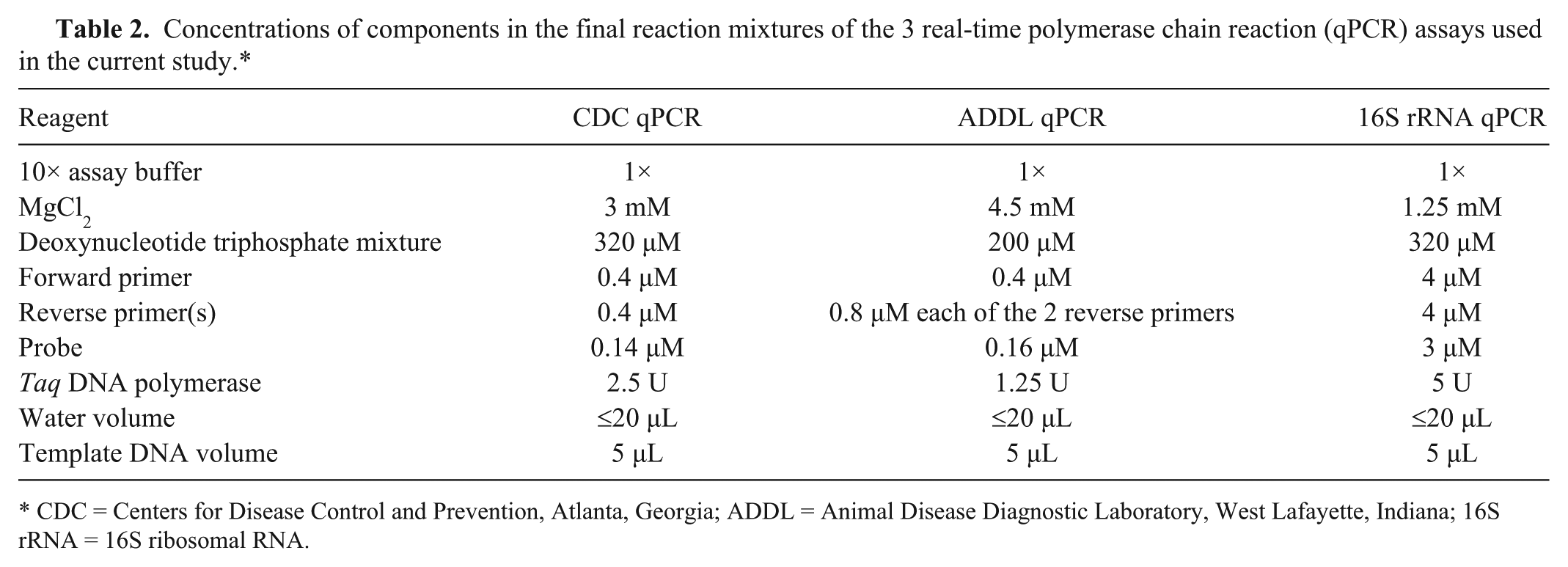
CDC = Centers for Disease Control and Prevention, Atlanta, Georgia; ADDL = Animal Disease Diagnostic Laboratory, West Lafayette, Indiana; 16S rRNA = 16S ribosomal RNA.
Gel-based PCR assay for pathogenic Leptospira detection
The previously described gel-based PCR assay 6 with minor modifications 18 was used. In this assay, specific DNA amplification results from 2 target regions that are specific to different groups of pathogenic Leptospira spp.; a positive amplification would result in either a 285-bp or a 563-bp fragment. The PCR assay was performed using the reagents from a commercial kit e and a thermocycler f as described earlier. 18 The amplified PCR products were separated via gel electrophoresis using a 1.5% agarose gel stained with ethidium bromide. The DNA bands were visualized and photographed under ultraviolet light.
Determination of analytical sensitivity
The lower limit of detection for each of the PCR assays was determined using genomic DNA extracted from L. interrogans serovar Pomona (obtained from the National Veterinary Service Laboratory, Ames, Iowa). The bacteria were grown at 30°C in complete Ellinghausen–McCullough–Johnson–Harris medium with 2% rabbit serum, and maintained by subculturing every 7–10 days. Enumeration of the bacteria was performed using dark-field microscopy and a Petroff–Hausser counting chamber. Freshly grown cultures at 1.2 × 108 leptospires/mL were used to prepare 10-fold serial dilutions by mixing 0.3 mL of culture or previous dilution with 2.7 mL of sterile, nuclease-free water in succession until a final dilution of 1.2 × 100 leptospires/mL was reached. Genomic DNA was extracted (see below for details) from each dilution and used as a template in the PCR assays. Standard curves for each qPCR assay were constructed using commercial software. g The average Ct values of 3 duplicate runs of each serial dilution for each qPCR assay were used for plotting the standard curves, and determining the linear relationship.
DNA extraction
Bacterial DNA was extracted from the culture serial dilutions as well as canine urine samples using a commercial kit. h One milliliter of each dilution or urine sample was transferred to a microcentrifuge tube, and centrifuged at 17,860 × g for 15 min at 4°C. The supernatant was removed, and the pellet was washed with 1 mL of sterile, nuclease-free water by centrifuging for another 15 min at 17,860 × g. The pellet was then resuspended in 160 µL of sterile, nuclease-free water, and 200 µL of ATL buffer h and 40 µL of proteinase K were added to the suspension. The DNA was extracted in 200 µL of buffer AE h as per the manufacturer’s suggested procedure and stored at −20°C until used for PCR assays. Five microliters of the extracted DNA was used as the template in each PCR assay. With each PCR run, DNA extracted from L. interrogans serovar Pomona culture was used for positive control, and nuclease-free water was used as negative control.
Sensitivity and specificity of the PCR assays
The sensitivity and specificity of the PCR assays in the detection of only pathogenic Leptospira strains was determined using genomic DNA isolated from cultures of pathogenic and nonpathogenic Leptospira strains (Table 3). Five microliters of each DNA sample was used as template in each PCR assay.
Genomic DNA of pathogenic and nonpathogenic Leptospira strains used for testing sensitivity and specificity of the real-time polymerase chain reaction assays used in the current study.*
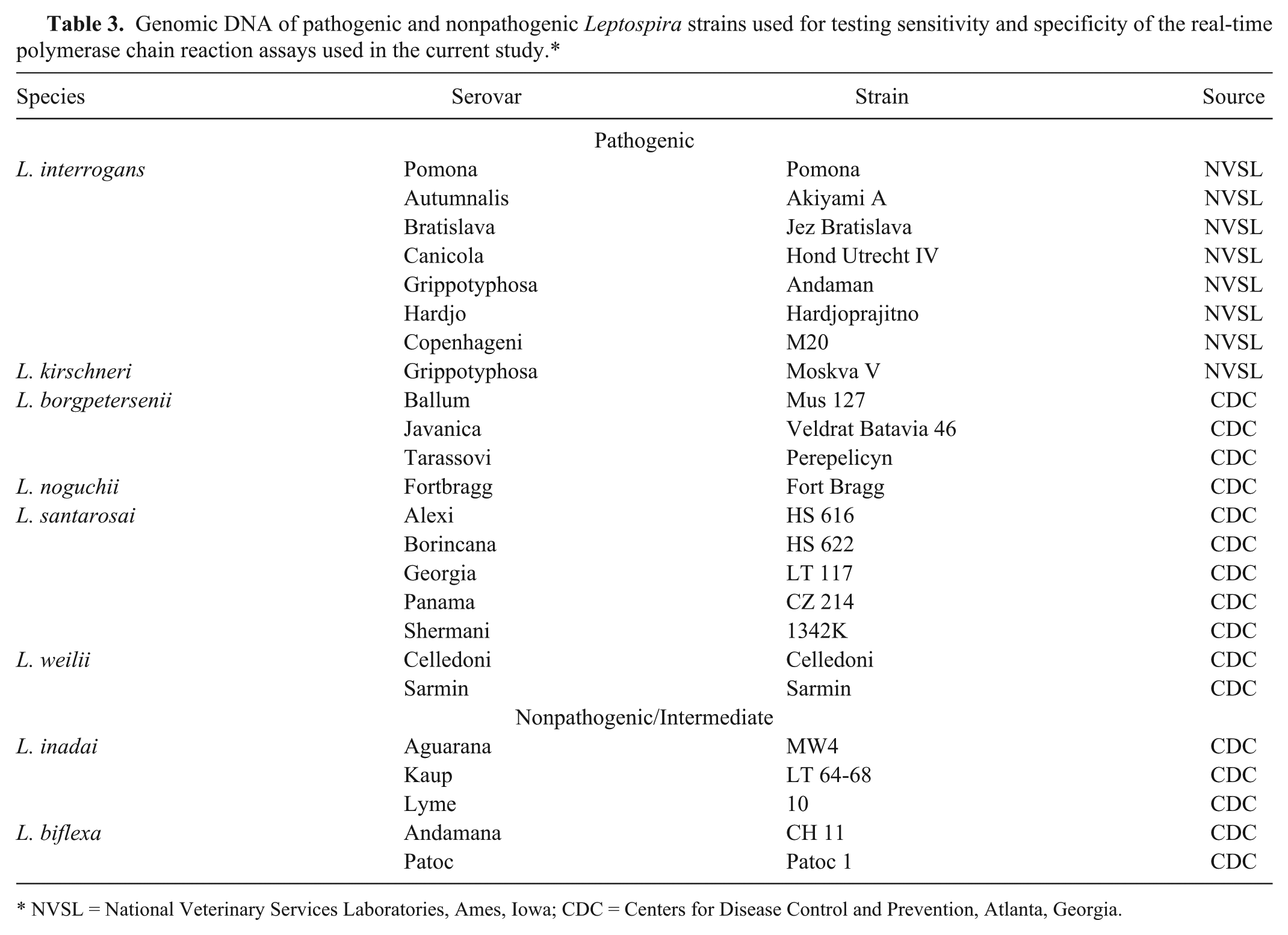
NVSL = National Veterinary Services Laboratories, Ames, Iowa; CDC = Centers for Disease Control and Prevention, Atlanta, Georgia.
Urine samples
Urine samples from 100 supposedly healthy dogs housed at the Humane Society of Indianapolis were obtained via the free-catch method over 2 months (May and June) in 2010. A plastic cup attached to a long rod was used to catch a mid-stream sample during the process of normal urination. The collected urine was transferred to a sterile polypropylene tube and transported on ice to the laboratory and processed immediately.
Urine samples from 42 dogs suspected of leptospirosis were collected during June–December of 2008–2011 by attending veterinarians at clinics in Indiana and Illinois participating in an affiliated project. The urine was removed aseptically via cystocentesis, transferred to a sterile polypropylene tube, and stored in a refrigerator until transported to the laboratory on ice. Extraction of DNA from the samples was usually initiated within 24–36 hr of urine collection. In most cases, the veterinarians also collected serum samples from the animals at the same time; the serum samples were submitted to the Indiana ADDL for MAT of antibody titers against 7 Leptospira serovars (Autumnalis, Bratislava, Canicola, Grippotyphosa, Hardjo, Icterohaemorrhagiae, and Pomona).
16S rRNA gene amplification and nucleotide sequence analysis
The prokaryotic 16S rRNA gene sequences were amplified from the DNA extracted from some urine samples collected from clinically ill dogs that were suspected to be suffering from leptospirosis. Consensus primers that are widely used for amplification of bacterial 16S rRNA gene sequences were used for this purpose (Table 1). Reagents from a commercial kit e and a thermocycler f were used for the amplification, with the following cycling parameters: 1 cycle at 95°C for 15 sec, followed by 34 cycles consisting of 30 sec at 93°C, 60 sec at 45°C, 60 sec at 72°C, and 1 cycle at 72°C for 7 min. The PCR products were then separated by electrophoresis on 1.5% agarose gel. The bands were visualized by staining with ethidium bromide.
The amplified 16S rRNA gene sequences and the amplified products of the 16S rRNA qPCR assay were cloned in a commercial system i using the protocol supplied by the manufacturer. The nucleotide sequences of the cloned fragments were determined at the Purdue Genomics Core Facility (West Lafayette, IN). The sequences were then compared to known bacterial sequences using the nucleotide BLAST program available from the National Center for Biotechnology Information website (http://www.ncbi.nlm.nih.gov/) to determine bacterial species.
Results
Performance of the 3 qPCR assays
All 3 qPCR assays detected DNA extracted from a range of 1.2 × 102 to 1.2 × 108 spirochetes/mL in a linear manner (Fig. 1). In the gel-based PCR assay, specific amplification was detected when the samples contained at least 1.2 × 104 spirochetes/mL; no specific amplification was detected in the samples with 1.2 × 103–1.2 × 100 spirochetes/mL (Fig. 2). When the qPCR assays were performed on 3 separate days using the same DNA samples, the interassay variability was found to be very low, with <0.5 differences in each Ct value. The 3 qPCR assays detected all of the 13 pathogenic, but not the intermediate and nonpathogenic, Leptospira strains tested (100% sensitivity and 100% specificity).
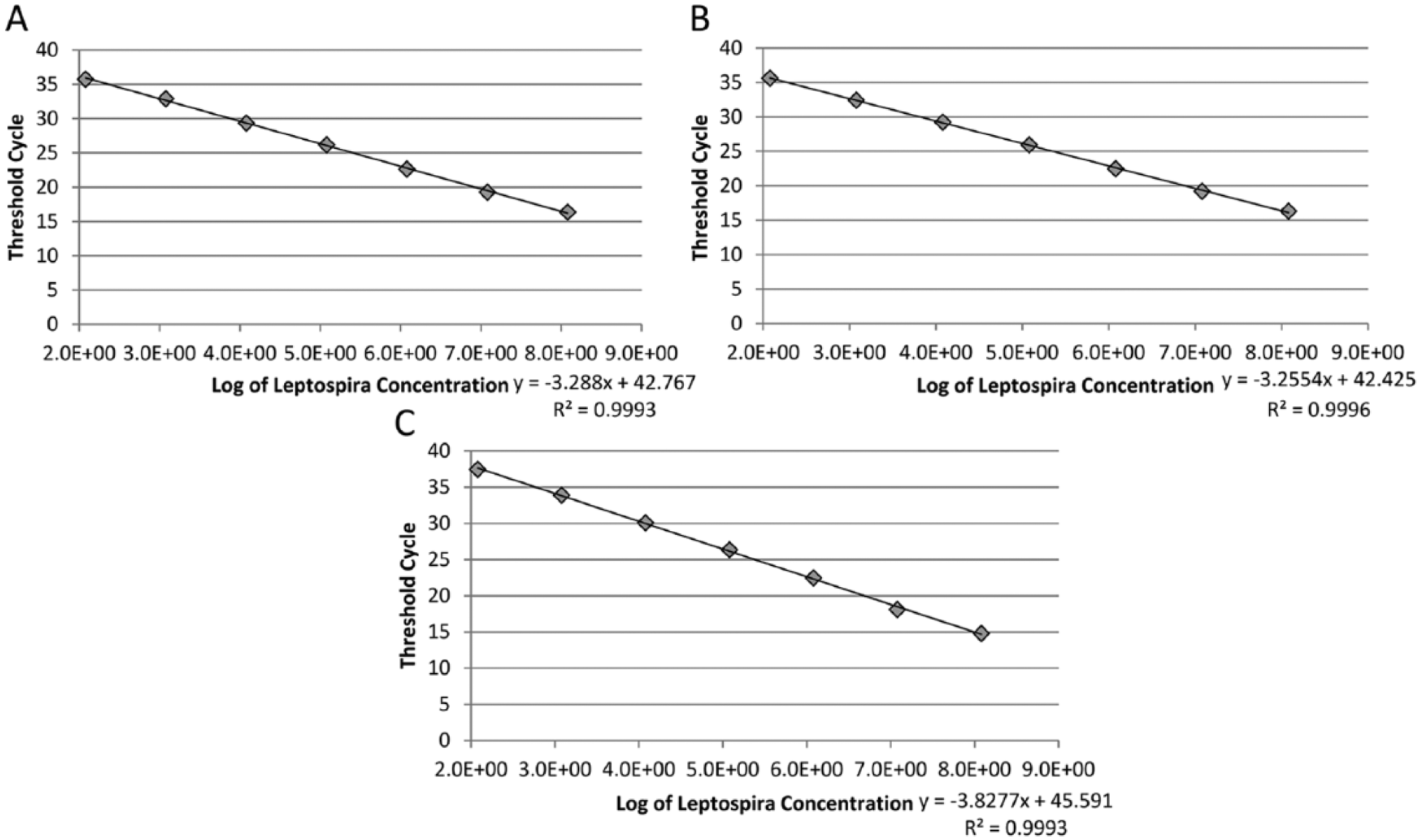
Analytical sensitivity of the Centers for Disease Control and Prevention (
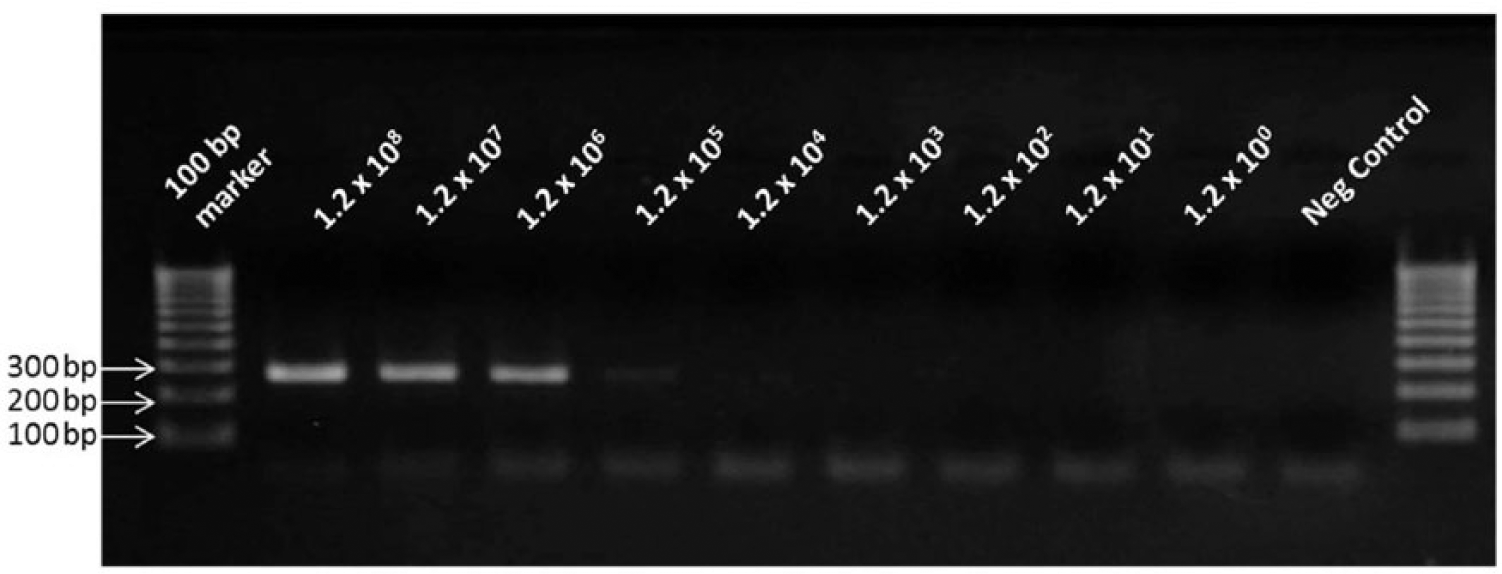
Detection sensitivity of the gel-based polymerase chain reaction. The assay detected a range of DNA extracted from 1.2 × 108 to 1.2 × 104 leptospires/mL. In the negative control, sterile, nuclease-free water was used in place of template DNA. Numbers at the left indicate the molecular size of the corresponding DNA fragment of the marker.
Evaluation of qPCR assays with urine samples from apparently healthy dogs
None of the 100 samples tested were positive for pathogenic Leptospira spp. by the CDC qPCR assay. Thirty of these 100 samples were randomly selected and tested using the ADDL qPCR and 16S rRNA qPCR assays. None of the 30 samples tested positive by the ADDL qPCR assay, but 4 out of 30 (13.3%) tested positive by the 16S rRNA qPCR assay, with Ct values ranging from 31.1 to 33.3. The 88-bp PCR products of positive samples were cloned, and their nucleotide sequences were determined. Nucleotide sequence analysis of the amplified fragments of the 4 positive samples revealed a 99–100% sequence identity with those from Leptospira spp. and several uncultured bacterial species from various environmental sources, including soil and animal skin surface (e.g., GenBank accession nos. EU534856, JN603762, JQ612372, JX631561, and KC453838).
Evaluation of qPCR assays with urine samples from clinically ill dogs
Aseptically collected urine samples from 42 clinically ill dogs were tested for pathogenic Leptospira spp. by the CDC qPCR assay (Supplementary Table 1). Two out of the 42 (4.8%) samples tested positive, with Ct values of 35.32 and 21.00, respectively. Thirty of the urine samples, including the CDC qPCR–positive samples, were tested by the ADDL qPCR and 16S rRNA qPCR assays and the results were in agreement with the CDC qPCR results. When the 2 positive samples were tested by the gel-based PCR assay, only the sample with a Ct value of 21 resulted in specific amplification (data not shown); the amplified product size was consistent with the presence of L. kirschneri. 18 To further confirm the infecting Leptospira species identity, the 16S rRNA gene was amplified from the extracted DNA, cloned, and sequenced. Nucleotide sequence analysis identified the infecting species as L. kirschneri with 100% sequence identity. The species identity of Leptospira in the other positive sample could not be determined because amplification of the 16S rRNA gene sequences was not obtained, most likely due to the low template DNA concentration in the sample.
Along with the urine samples, acute serum samples were also submitted for 35 dogs. Fourteen of these dogs (40.0%) tested negative by MAT for all 7 Leptospira serovars tested, while 11 dogs (31.4%) had a MAT titer of 100 or 200 against at least 1 of the serovars tested. Ten dogs (28.6%), including the 2 qPCR-positive animals, had a MAT titer of 400 or above (range: 400–6,400) against 1 of the serovars (Supplementary Table 1; available at jvdi.sagepub.com/supplemental). Convalescent serum samples were obtained for 3 of these dogs, and 2 of them had a 4-fold increase in the MAT titers compared to the acute serum sample, 1 (sample number 40) of which tested negative by all 3 qPCR assays (Supplementary Table 1).
In order to determine the presence of any bacterial pathogens in urine samples that tested negative for pathogenic Leptospira spp., the 16S rRNA gene sequences present in the extracted DNA were amplified. Out of the 15 tested, successful amplification was obtained from 4 samples (data not shown). Nucleotide sequence analyses of the amplified fragments identified the presence of Klebsiella sp., Pseudomonas sp., Escherichia coli, and Enterobacter sp. in each of the urine samples (data not shown).
Discussion
The 3 qPCR assays used in this study had similar analytical sensitivity and were able to consistently detect Leptospira DNA extracted from samples with the lowest concentration of 1.2 × 102 leptospires/mL, which is equivalent to DNA of 3 leptospires per PCR reaction. This level of sensitivity is comparable to the previously reported results with the CDC and 16S rRNA qPCR assays.16,18 Though both CDC and ADDL qPCRs targeted the lipl32 gene, there is a difference with regard to the primer and probe sequences and the amplified DNA product size. The ADDL qPCR amplified a 76-bp fragment while the CDC qPCR amplified a 241-bp fragment. It is generally considered that amplification of smaller sized DNA fragments is much more efficient than the larger sized ones. 2 The 241-bp PCR amplicon of the CDC qPCR is longer than the recommended 50–150-bp size for qPCR assays. 13 However, the results of the current study showed no difference between the ADDL and CDC qPCR assays in terms of the sensitivity and specificity or Ct values for each of the template DNA concentrations. As expected, the qPCR assays were at least 100-fold more sensitive than the gel-based conventional PCR. In addition to better sensitivity, qPCR assays have several advantages compared to the gel-based PCR assays, including a shorter turnaround time, increased specificity when 5′ nuclease assay probes are used, as well as a substantially less likelihood of cross-contamination. 11
All 3 qPCR assays detected the pathogenic, but not the intermediate or saprophytic, Leptospira spp. tested in this study. The sensitivity and specificity of the CDC and ADDL qPCR assay is not surprising as primers and probes of these assays were specific to the lipl32 gene that is present only in pathogenic Leptospira spp. 17 On the other hand, the 16S rRNA qPCR amplifies the highly conserved 16S rRNA gene, and the primers and probe for this assay were designed for specific amplification from pathogenic Leptospira spp. as their nucleotide sequences were significantly different from those of all other bacterial species available in the databases in 2002. 16 However, the results of the current study with the free-catch urine samples demonstrate that the 16S rRNA qPCR can detect DNA of certain commensal or environmental bacteria. When the current study found that some urine samples from healthy dogs were positive for pathogenic Leptospira spp. by the 16S rRNA qPCR, but not by the CDC or ADDL qPCR assays, it was hypothesized that the 16S rRNA qPCR assay was probably detecting the presence of some other bacterial species. The nucleotide sequence analysis of the amplified DNA fragment confirmed that this assay can also amplify DNA of some bacterial species that are present on normal skin surface or in the environment. The false-positive results with the 16S rRNA qPCR assay were only observed when urine samples were collected by free-catch method; no false positives were seen with collection by cystocentesis. It is possible that during free catch, bacteria present on skin and hair of dogs could contaminate the collected urine sample. Also, any soil contamination of the collection cup could lead to the presence of environmental bacteria in the urine leading to false-positive results in the 16S rRNA qPCR assay. The high level of nucleotide sequence identity between the amplified regions of Leptospira spp. and the commensal and/or environmental bacteria makes this assay unsuitable for diagnostic application.
None of the 100 shelter dog urine samples tested positive for pathogenic Leptospira spp. It has been reported that as many as 8.2% of the apparently healthy dogs in the United States shed pathogenic Leptospira spp. in their urine using a gel-based PCR that amplified a portion of the conserved 23S rRNA gene. 8 A study in Ireland, using a qPCR to amplify the lipl32 gene similar to the one used in the current study, reported the shedding of pathogenic Leptospira in urine of 7.05% of healthy canines. 14 The discrepancy between the results of the current study and that of the other 2 previous studies could be because of several reasons, including differences in sensitivity and specificity of the assays, annual changes in prevalence of canine leptospirosis, different prevalence of pathogenic serovars (e.g., serovar Canicola), and different geographical location and health status of the dogs. It is also possible that shedding of pathogenic Leptospira in urine of healthy dogs is not highly prevalent.
Only 2 of the 42 samples from dogs that attending veterinarians suspected of leptospirosis were positive for pathogenic Leptospira spp. The low number of positive samples could be due to the transient and/or intermittent shedding by dogs of the pathogenic Leptospira in urine. 7 This might also be the reason why a sample (no. 40) tested negative by qPCR, even though the 4-fold increase in the MAT titer of its convalescent serum suggested an active Leptospira infection. Shedding a low number of organisms that was below the limit of detection for the qPCR assays could be another reason for the negative test with this sample. It is possible that some of the dogs that tested negative for Leptospira spp. were truly negative and that their clinical disease was caused by other pathogens. In fact, the limited effort to detect the presence of other bacterial pathogens in the urine identified 4 bacteria (Klebsiella, Pseudomonas, Enterobacter, and E. coli) that are known to cause urinary tract infections. 15
Generally, the specificity of PCR assays is determined by in silico analysis (i.e., BLAST search of nucleotide sequence databases) followed by testing with nucleic acids extracted from closely related organisms as well as other clinically relevant organisms. 4 The results of the current study highlight the importance of sample-specific validation of PCR-based diagnostic assays and the application of appropriately validated assays for more reliable pathogen detection. The 16S rRNA qPCR assay is unsuitable for routine detection of Leptospira spp. in clinical samples that are contaminated with commensal and/or environmental bacteria, such as urine collected by free-catch. In view of the equivalent assay performances, and the fact that the ADDL qPCR requires the use of 2 reverse primers, it is suggested that the CDC qPCR is well suited for routine use in the detection of pathogenic Leptospira spp. in canine urine samples.
Footnotes
Acknowledgements
The authors thank the Humane Society of Indianapolis and all the veterinary clinicians who submitted urine and serum samples from dogs suspected for leptospirosis. The authors also thank Cecilia Santrich for her laboratory technical help, and Dr. Robyn Stoddard, Centers for Diseases Control and Prevention, for providing genomic DNA of several Leptospira strains.
a.
Beacon Designer, PREMIER Biosoft International, Palo Alto, CA.
b.
Eurofins MWG Operon, Huntsville, AL.
c.
Platinum Taq DNA polymerase, Life Technologies, Carlsbad, CA.
d.
Smart Cycler II, Cepheid Inc., Sunnyvale, CA.
e.
NovaTaq Hot Start DNA polymerase kit, EMD Millipore Corp., Billerica, MA.
f.
Hybaid MultiBlock thermal cycler system, Thermo Scientific Hybaid, Milford, MA.
g.
EXCEL, Microsoft Corp., Redmond, WA.
h.
ATL buffer, DNeasy blood and tissue kit; Qiagen Inc., Valencia, CA.
i.
pGEM T Easy vector system, Promega Corp., Madison, WI.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported in part by a grant from the Morris Animal Foundation.